Indice del volumen Volume index
Comité Editorial Editorial Board
Comité Científico Scientific Committee
- Microscopia de luz
En varones con SALX y en las mujeres portadoras los cambios son mínimos al inicio de la enfermedad, posteriormente hay aumento leve de la celularidad del mesangio, que puede ser focal o segmentaria. En estados más avanzados de la enfermedad se puede encontrar glomeruloesclerosis segmentaria o difusa, acompańado de atrofia tubular y fibrosis intersticial junto con células espumosas2-4.
- Inmunofluorescencia
Se pueden encontrar depósitos no específicos de IgG, IgM y fibrinógeno, además de IgA, C3, C4 y C1q, aunque en menor grado. La inmunofluorescencia para colágeno tipo IV en piel o biopsia renal puede ser útil para hacer el diagnóstico.
En pacientes con SALX, las cadenas
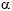 3 (IV), 4 (IV) y 5 (IV) de colágeno en la mayoría de los glomérulos, túbulos y cápsula de Bowman son indetectables por inmunohistoquímica, pero sí expresan las cadenas 1 (IV) y 2 (IV). Las mujeres heterocigotas frecuentemente muestran tramos de la MBG que fallan en la expresión de las cadenas 3 (IV), 4 (IV) y 5 (IV). En pacientes con SAAR, existe colágeno tipo 5 (IV) en la cápsula de Bowman, pero no se identifican las cadenas de colágeno 3 (IV) y 4 (IV) en la MBG, en la membrana basal tubular, o en la de epidermis. La razón de esta diferencia estriba en que la cadena de colágeno 5 (IV) se encuentra compartida por dos protómeros (uno en la MBG y otro en la cápsula de Bowman)2-4.
3 (IV), 4 (IV) y 5 (IV) de colágeno en la mayoría de los glomérulos, túbulos y cápsula de Bowman son indetectables por inmunohistoquímica, pero sí expresan las cadenas 1 (IV) y 2 (IV). Las mujeres heterocigotas frecuentemente muestran tramos de la MBG que fallan en la expresión de las cadenas 3 (IV), 4 (IV) y 5 (IV). En pacientes con SAAR, existe colágeno tipo 5 (IV) en la cápsula de Bowman, pero no se identifican las cadenas de colágeno 3 (IV) y 4 (IV) en la MBG, en la membrana basal tubular, o en la de epidermis. La razón de esta diferencia estriba en que la cadena de colágeno 5 (IV) se encuentra compartida por dos protómeros (uno en la MBG y otro en la cápsula de Bowman)2-4.
- Microscopia electrónica
Las alteraciones consisten en áreas de adelgazamiento irregular y otras de engrosamiento variable, difuso o focal de la MBG, con un borde subepitelial irregular y complejo, con una replicación entretejida de la lámina densa, resultando en aspecto hojaldrado. Las áreas claras que se observan entre las ramas de la lámina densa a menudo contienen partículas densas. En etapas tempranas de la enfermedad (nińos y mujeres portadoras de la forma ligada a X), la membrana basal sólo puede parecer adelgazada. Los hallazgos ultraestructurales no necesariamente correlacionan con un tipo de mutación y puede haber diferencias aún en la misma familia2-5.
- Análisis de ligamiento (indirecto)
El análisis de ligamiento está totalmente basado en el diagnóstico clínico, por lo que es de vital importancia disponer de una historia clínica lo más completa posible de la familia que debe estudiarse. Este análisis permite identificar el patrón de herencia de una enfermedad mediante el establecimiento del haplotipo de riesgo en una familia1-5. - Análisis mutacional (directo)
Los genes del colágeno tipo IV son de gran longitud (una media de 50 exones por gen) y con una gran complejidad, que dificulta su estudio exhaustivo para la detección de la mutación causante de la enfermedad. No existe ninguna técnica rápida aplicable en el ámbito asistencial para el diagnóstico mutacional del SAAR, mientras que el estudio de ARN a partir de raíz de cabello permite el diagnóstico mutacional del gen COL4A5. - Fase I: Abarca desde el nacimiento hasta la infancia tardía o adolescencia temprana. La única alteración clínica es la hematuria, con cambios histopatológicos limitados a una proliferación mesangial y adelgazamiento de la MBG, con áreas focales de laminación y podocitos normales.
- Fase II: Estos pacientes presentan proteinuria además de la hematuria, pero la velocidad de filtración glomerular se mantiene aún normal. En la biopsia renal se encuentra proliferación mesangial y puede haber áreas con esclerosis focal y segmentaria; el área tubulointersticial se encuentra intacta. Por microscopia electrónica ya se pueden identificar engrosamiento difuso y laminación de la MBG con áreas extensas de fusión de pedicelos.
- Fase III: Se caracteriza por deterioro en la función renal; en los hallazgos histopatológicos existe dańo intersticial y atrofia tubular.
- Fase IV: Los pacientes manifiestan datos clínicos de enfermedad renal terminal.
- Inhibidores de enzima convertasa (IECA):
En un modelo canino de SA ligado al cromosoma X, el tratamiento con IECA tuvo efectos benéficos en la función renal, estructura renal y supervivencia. En la mayoría de los pacientes tratados con IECA, hay una disminución inicial en la velocidad de filtración glomerular, que se explica por los efectos hemodinámicos del fármaco. Proesmans y Van Dyck reportaron a 10 nińos con SA y proteinuria igual o mayor de 350 mg/24 horas, con velocidad de filtración glomerular por arriba de 80 mL/ min/1.73 m2, que recibieron enalapril por cinco ańos; observaron disminución en la proteinuria y preservación de la velocidad de filtración glomerular; sin embargo, no se trató de un estudio aleatorizado comparativo.
- Ciclosporina:
En casos de síndrome nefrótico, varios grupos han utilizado ciclosporina. Este es un fármaco que actúa inhibiendo la acción de la calcineurina. Tiene un efecto antiproteinúrico en varias glomerulopatías, tanto humanas como experimentales, y se ha sugerido que el reducir la proteinuria puede retardar la progresión del dańo renal; sin embargo, se debe tener cuidado con el uso prolongado por el efecto nefrotóxico del medicamento.
- Espironolactona:
En un estudio de cinco pacientes con SA, que continuaban con proteinuria a pesar del tratamiento con IECA o con antagonistas de receptores de angiotensina, Kaito y col. agregaron espironolactona como antagonista de receptor de mineralocorticoides; después de un seguimiento a 18 meses observaron reducción significativa en la proteinuria con esta combinación, sin deterioro en la función renal o hiperkalemia.
- Inhibidores de 3-hidroxi-3-metaglutaril coenzima A reductasa (HMG-CoA).
Se ha reportado que los hipolipemiantes inhibidores de la HMG-CoA reductasa, enzima que cataliza la conversión de HMG-CoA a mevalonato, paso inicial limitante en la síntesis del colesterol, tienen efectos pleiotrópicos, por los que modulan la producción de tejido conectivo; Koepke y col. realizaron un estudio en un modelo murino, en el que se anuló la expresión del gen COL4A5 (knockout), usando como tratamiento cerivastatina, encontrando que esta terapia prolonga la vida de los ratones, reduce la proteinuria y retarda la uremia. Estos efectos se asociaron a disminución en la fibrosis renal y en el infiltrado inflamatorio por células T, macrófagos y miofibroblastos en el tejido renal8-11. - Trasplante de células madre.
Prodromidi y col. 12realizaron un estudio en ratones knockout para COL4A5 que recibieron trasplante de médula ósea de ratones normales (salvajes o Wild type), encontrando que los ratones trasplantados tuvieron expresión de colágena3 en el glomérulo por inmunofluorescencia; el ARN mensajero de dicha proteína se pudo detectar por PCR, y concluyen que estos alentadores resultados se pueden deber a generación de podocitos sin el defecto genético8-11, 13.
SÍNDROME DE ALPORT.
II.- DIAGNÓSTICO Y TRATAMIENTO
Natalia Zabala MD.
Sanatorio de la Trinidad Palermo.
Buenos Aires, Argentina
natalialzabala @ hotmail.com
Rev Electron Biomed / Electron J Biomed 2016;2:37-45.
Comentario del revisor Prof. Dr. Juan F. Macías Núńez. Servicio de Nefrología. Hospital Universitario de Salamanca. Espańa
Comentario del revisor Dra. Mariana Ciocchini. Instituto Daomi - Facultad de Medicina (UBA). Universidad de Buenos Aires, Buenos Aires, Argentina
RESUMEN
El diagnóstico del síndrome de Alport puede basarse en diversas medidas terapéuticas que oscila desde los inhibidores de enzima convertasa, ciclosporina, espironolactona, el trasplante renal y hasta tratamientos en fase experimental, tales como los inhibidores de 3-hidroxi-3-metaglutaril coenzima A reductasa o el trasplante de células madres.
PALABRAS CLAVE: Asíndrome de Alport, diagnóstico, tratamiento.
SUMMARY:
Alport syndrome diagnose is based on a combination of clinical, genetic and histopathologic data. Treatment may be based on various therapeutic measures ranging from inhibitors of convertase enzyme, cyclosporine, spironolactone, kidney transplantation
KEY WORDS: Alport syndrome, diagnosis, treatment.
INTRODUCCIÓN
En la primera parte de este artículo se ha desarrollado todo lo referido a su fisiopatología y manifestaciones clínicas. En esta segunda parte se desarrollará lo referido a su diagnóstico y tratamiento.
DIAGNÓSTICO
Criterios diagnósticos generales
En el caso que existan antecedentes familiares, para que un paciente sea diagnosticado definitivamente de Síndrome de Alport (SA) debe cumplir, al menos, dos de los siguientes criterios diagnósticos y al menos cuatro si no existen dichos antecedentes:
1. Historia familiar de nefropatía o hematuria idiopática en un familiar de primer grado del paciente índice o en un familiar hombre emparentado con él a través de cualquier número de generaciones de mujeres.
2. Hematuria persistente sin la evidencia de cualquier otra posibilidad de nefropatía hereditaria, como la enfermedad de la membrana basal delgada, poliquistosis renal o la nefropatía por IgA.
3. Hipoacusia bilateral neurosensorial en el rango de 2.000-8.000 Hz. La hipoacusia se desarrolla gradualmente, no está presente en la infancia temprana y suele presentarse antes de los 30 ańos.
4. Una mutación en COL4An
5. Evidencia inmunohistoquímica de ausencia parcial o total del epítopo de Alport en la MBG, en la membrana basal epidérmica (MBE), o en ambas.
6. Anomalías ultraestructurales repartidas por toda la MBG, en particular, engrosamiento, adelgazamiento y lamelación.
7. Lesiones oculares entre las que se incluyan: lentícono anterior, catarata subcapsular posterior, distrofia polimórfica posterior y flecos retinianos.
8. Progresión gradual a IRCT en el paciente índice o como mínimo en dos de los miembros de su familia.
9. Leiomiomatosis difusa del esófago, genitales femeninos o ambas.
Biopsia Renal: Diagnostico Histopatológico:
El procedimiento diagnóstico habitual que confirma la sospecha clínica es la biopsia renal1-5.
Actualmente, debido a que la cadena del colágeno 5 (IV) también está presente en la MBE, se están realizando estudios de inmunohistoquímica en biopsias de piel como procedimiento poco invasivo y alternativo a la biopsia renal.
En la mayor parte de las biopsias de piel realizadas a hombres con SALX, se ha comprobado que existe una ausencia completa de esta cadena; sin embargo, en las mujeres portadoras de SALX se ha observado un mosaicismo en la expresión de la cadena 5 (IV) en la piel (dos tercios de las mujeres portadoras presentan un patrón inmunohistoquímico anormal), lo que concuerda con el fenómeno de inactivación del cromosoma X que puede ser diferente en cada una de sus células.
No hay acuerdo sobre si la mayor o menor expresión de las cadenas 5 (IV) en la piel, se correlaciona en mujeres con la gravedad de la afectación renal1-5.
Diagnóstico Molecular
Debe tenerse en cuenta la historia familiar de cada paciente índice para utilizar mejor las técnicas de biología molecular.
Aproximadamente un 10% de las mutaciones en el gen COL4A5 son de novo, pero está por demostrarse qué porcentaje de estos casos pueden deberse a mosaicismo germinal/genosómico en los progenitores.
Es muy importante el diagnóstico de portadoras de SALX. Por lo tanto, ante el diagnóstico de un hombre-caso índice debe procederse a descartar que las mujeres sean portadoras. Este diagnóstico tiene trascendencia de cara a que estas mujeres presentan el riesgo de tener hijos afectos, deben ser seguidas en una consulta de nefrología y debe considerarse que no son idóneas como donantes.
El diagnóstico de SALX en familias en las que no hay hombres afectados resulta difícil y, si no se piensa en esta posibilidad, las familias pueden ser etiquetadas como afectadas de hematuria familiar, con el riesgo de que una de las mujeres llegue a tener un hijo afectado de SALX1,6.
EVOLUCIÓN
En los últimos ańos se ha puesto en claro que las mujeres con SA están en riesgo de progresar a insuficiencia renal crónica, contrario a lo que se pensaba antes. Este riesgo se incrementa con la edad; así, 41% de las mujeres con SA que se encuentren entre la cuarta y sexta décadas de la vida desarrollarán insuficiencia renal crónica terminal3-5.
Dentro de los factores de riesgo que se han encontrado, destacan el antecedente de haber tenido hematuria macroscópica en la infancia, síndrome nefrótico y engrosamiento difuso de la MBG en la microscopia electrónica.
Estas observaciones tienen implicaciones clínicas muy importantes para el personal médico en el seguimiento de estas familias y para la selección de donadores relacionados en los pacientes con SA que requieren de un trasplante renal.
Historia natural de la nefropatía por SA
El dańo renal del SA puede dividirse en varias fases5:
El tiempo estimado en que un paciente puede pasar de una fase a otra dependerá del tipo de mutación que presente. Pacientes en los que la producción de cualquier proteína funcional se encuentra frenada (deleciones y mutaciones sin sentido) pasan a través de estas fases más rápido que aquéllos en los que las mutaciones permiten una síntesis funcional de una proteína (mutaciones de codón)3,5
TRATAMIENTO
No existen estudios clínicos aleatorizados de tratamiento de la nefropatía en el SA.
En el SALX, todos los varones afectados desarrollan uremia terminal; la proporción de mujeres con falla renal varía según el tipo de SA5,6.
Tanto en hombres como en mujeres, la proteinuria y la hipertensión son los mayores indicadores de progresión de dańo renal; por esto, muchos grupos han encaminado el esfuerzo en reducir la proteinuria con fármacos hipotensores6,7:
Trasplante renal
Actualmente, el trasplante renal constituye el único tratamiento eficaz para los pacientes con SA. Un 3-4% de los hombres sometidos a trasplante desarrollan una glomerulopatía por anticuerpos anti-MBG. Este hecho es más frecuente en paciente portadores de mutaciones de cambio de pauta de lectura o deleciones. El dańo tisular está mediado por autoanticuerpos que se unen a la membrana basal glomerular. Kalluri, et al. han descripto que todos los pacientes con SA sometidos a trasplantes desarrollan una respuesta inmunitaria humoral contra las cadenas 3(IV), 4(IV) y 5(IV). Sin embargo, no todos desarrollan un síndrome de Goodpasteur, por lo que se cree que el desarrollo de éste debe depender de la capacidad de cada individuo de poner en marcha una respuesta celular contra la cadena 3 (IV) que previamente no poseían o no expresaban y, por lo tanto, se comporta como un nuevo epítopo antigénico7-8.
Existe una cierta controversia referente al hecho de si una mujer portadora de SALX puede o no ser donante. Teniendo en cuenta que una gran parte de estas mujeres desarrollarán proteinuria y que una parte no despreciable (hasta un tercio a los 60 ańos) llegarán a alcanzar IRCT, deberían desestimarse como donantes. Sólo en casos que a los 40-50 ańos no presenten ni microalbuminuria ni hipoacusia podría valorarse que fuesen donantes, pero siempre sería tras haber descartado a otros posibles donantes7-8.
Tratamientos en experimentación
La disponibilidad de modelos animales de SA, tanto murinos como caninos, ha permitido realizar estudios de tratamiento que en un futuro podrán aplicarse a seres humanos; entre éstos destacan7.:
CONCLUSIÓN:
El síndrome de Alport es una enfermedad hereditaria, usualmente ligada al cromosoma X, que se caracteriza por presentar nefropatía progresiva, sordera y oftalmopatía, y se asocia a una alteración en el colágeno tipo IV.
REFERENCIAS
-
1.- Pirson Y. Making the diagnosis of Alport's syndrome. Kidney Int 1999;56(2):760-75
2.-) Kashtan C. Alport syndrome: facts and opinions.F1000Res. 2017 Jan 17;6:50. doi: 10.12688/f1000research.9636.1. eCollection 2017
3.- Kashtan CE. Alport Syndrome and Thin Basement Membrane Nephropathy. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Bird TD, Ledbetter N, Mefford HC, Smith RJH, Stephens K, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2017.2001 Aug 28 [updated 2015 Nov 25
4.- Deltas C, Savva I, Voskarides K, Papazachariou L, Pierides A.Carriers of Autosomal Recessive Alport Syndrome with Thin Basement Membrane Nephropathy Presenting as Focal Segmental Glomerulosclerosis in Later Life. Nephron. 2015;130(4):271-8
5.- Kashtan CE. Alport syndrome: abnormalities of type IV collagen genes and proteins. Ren Fail 2000;22(6):737-49
6.- Kashtan CE. Alport syndrome and the X chromosome: implications of a diagnosis of Alport syndrome in females. Nephrol Dial Transplant 2007;22(6):1499-505
7.- Cosgrove D, Liu S.Collagen IV diseases: A focus on the glomerular basement membrane in Alport syndrome. Matrix Biol. 2017;57-58:45-54
8.- Gross O, Weber M, Fries JW, Muller GA. Living donor kidney transplantation from relatives with mild urinary abnormalities in Alport syndrome: long-term risk, benefit and outcome. Nephrol Dial Transplant 2009;24(5):1626-30.
9.-) Byrne MC, Budisavljevic MN, Fan Z, Self SE, Ploth DW. Renal transplant in patients with Alport's syndrome. Am J Kidney Dis 2002;39(4):769-75
10.- Mochizuki T, Lemmink HH, Mariyama M, et al. Identification of mutations in the alpha 3(IV) and alpha 4(IV) collagen genes in autosomal recessive Alport syndrome. Nat Genet 1994;8(1):77-81
11.- Savva I, Pierides A, Deltas C. RAAS inhibition and the course of Alport syndrome. Pharmacol Res. 2016;107:205-10
12.- Prodromidi EI, Poulsom R, Jeffery R, Roufosse CA, Pollard PJ, Pusey CD, Cook HT.Bone marrow-derived cells contribute to podocyte regeneration and amelioration of renal disease in a mouse model of Alport syndrome. Stem Cells. 2006;24(11):2448-2455.
13.- Zhang Y, Wang F, Ding J, Zhang H, Liu X, Wang S, Xiao H, Yao Y, Liu J, Zhong X, Guan N, Su B, Wu G, Yu L.Long-term treatment by ACE inhibitors and angiotensin receptor blockers in children with Alport syndrome.Pediatr Nephrol. 2016;31(1):67-72
CORRESPONDENCIA:
Dra. Natalia Zabala
Sanatorio de la Trinidad Palermo.
Buenos Aires
Argentina
Email: natalialzabala @ hotmail.com
Comentario del revisor Prof. Dr. Juan F. Macías Núńez. Servicio de Nefrología. Hospital Universitario de Salamanca. Espańa
En la glomerulopatía del Síndrome de Alport se ha demostrado un incremento significativo de la podocituria con respecto a los valores hallados en la pobla-ción sana, lo cual condice con la hipótesis de la depleción podocitaria como mecanismo fisiopatológico común a las glomerulopaías.
Comentario del revisor Dra. Mariana Ciocchini. Instituto Daomi - Facultad de Medicina (UBA). Universidad de Buenos Aires, Buenos Aires, Argentina
Los fármacos inhibidores de la enzima convertidora de la angiotensina II estabilizan al podocito reduciendo la podocituria y, actualmente, se recomienda emplearlos en pacientes con Síndrome de Alport aun antes de que presente pro-teinuria o hipertensión
Referencias:
1. Wickman L, Farsad Afshinnia F, Su Q. Wang SQ et al. Urine Podocyte mRNAs, Pro-teinuria, and Progression in Human Glomerular Diseases. J Am Soc Nephrol 2013; 24: 2081-2095.
2. Gross O, Friede T, Hilgers R et al. Safety and Efficacy of the ACE-Inhibitor Ramipril in Alport Syndrome: The Double-Blind, Randomized, Placebo-Controlled, Multicenter Phase III EARLY PRO-TECT Alport Trial in Pediatric Patients. ISRN Pediatr 2012:436-46.
3. Savige J, Gregory M, Gross O et al. Expert Guidelines for the Management of Alport Syndrome and Thin Basement Membrane Nephropathy. J Am Soc Nephrol 2013; 24: 364-375.