Indice del volumen Volume index
Comité Editorial Editorial Board
Comité Científico Scientific Committee
VARIANTE TRICOLEUCÉMICA DE CÉLULAS B, CON RESTRICCIÓN ISOTÍPICA KAPPA Y AFECTACIÓN DÉRMICA.
INFORME DE UN CASO.
Dr. Ernesto Moro-Rodríguez1, Inmaculada de Prada-Vicente2, Montserrat Pérez 4,
Carmen Alonso-San Pablo 1, y Miguel Ángel Martínez-González3
1Universidad Rey Juan Carlos, Facultad de Ciencias de la Salud, Alcorcón, 2Hospital Materno-Infantil Nińo Jesús,
3Hospital 12 de Octubre (Madrid). 4Hospital Virgen de la Concha (Zamora). Espańa
Rev Electron Biomed / Electron J Biomed 2004;3:27-32.
Comentario del Revisor Dr. Oscar Marín. Hospital Pablo Soria de Jujuy. San Salvador de Jujuy. Argentina
Comentario del Revisor Dr. Bayardo Flores. Istituto Cantonale di Patologia. Locarno. Suiza
Comentario del Revisor Prof. Reynaldo Falcón-Escobedo Facultad de Medicina, Universidad Autónoma de San Luis Potosí, San Luis Potosí, S.L.P. México
Palabras clave: Proceso linfoproliferativo, linfocitos B, Variante Tricoleucémica, lesiones cutáneas, microscopía electrónica.
Introducción
La Tricoleucemia o Leucemia de Células Peludas (LCP), es una enfermedad linfoproliferativa crónica, caracterizada por esplenomegalia, pancitopenia e infecciones recurrentes. Desde la descripción de Bouroncle en 1958 se describe que está caracterizada por una población de células cuyo rasgo más característico es que poseen prolongaciones citoplasmáticas a modo de "pelos" (tricoleucocitos). Estas células suelen presentar una fuerte reacción fosfatasa ácida tartrato resistente y casi siempre tienen un inmunofenotipo B. Este tipo de proceso representa entre el 1 al 2% de todos los síndromes linfoproliferativos crónicos y suelen afectar a varones con una proporción 4/1.
Al margen de éste cuadro clásico se ha descrito la que se conoce como Variante Tricoleucémica, que afecta fundamentalmente a mujeres, suelen tener células tartrato resistente negativas, presentan una menor fibrosis en médula ósea y además, a nivel ultraestructural los elementos celulares carecen de complejos ribosómicos laminares ("lamelares") característicos de la tricoleucemia clásica.
Dentro de lo inusual de las tricoleucemias variantes, el caso presentado debutó con una infiltración dérmica de ambas mamas comprometiendo su areola, hecho informado infrecuentemente. En la exploración física completa de la paciente se puso de manifiesto una enfermedad sistémica con presencia de esplenomegalia gigante, hepatomegalia, derrame pleural e infiltración de la médula ósea
INFORME DEL CASO.
Mujer de 74 ańos de edad a la que se le atiende en interconsulta por presentar lesiones purpúreas de 15 meses de evolución localizadas en ambas mamas que adoptaban una forma en llamarada y que afectaban al pezón y a la areola (figura 1). La paciente refirió que se habían iniciado y desarrollado de forma progresiva en ambas mamas y que dada la ausencia de dolor y de supuración por los pezones no sintió la necesidad de consultarlo. El cuadro clínico se acompańaba desde hacía un ańo de anorexia y pérdida de peso de 20 Kg. Quince días antes de su ingreso comenzó con disnea que se hizo progresiva, motivo por el que acudió a Urgencias.
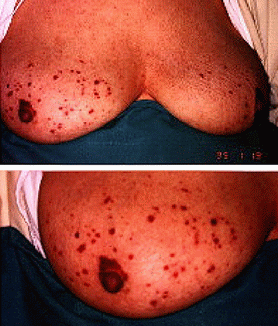
Figura 1
De la exploración física destacaba una disminución del murmullo vesicular en mitad inferior del hemitórax derecho; un abdomen globuloso con esplenomegalia gigante que llegaba hasta la fosa ilíaca, en límite línea media umbilical, no dolorosa; y hepatomegalia a 7 cm. del reborde costal. No se palparon adenopatías periféricas clínicamente significativas.
Datos complementarios más destacados:
HEMOGRAMA: leucocitos 2200 (27 S, 1C, 68 L, 2 M, 2 Eo), Hb 12.5, Hematocrito 37.8, VCM 98, ADE 15.7, Plaquetas 125.000; reticulocitos 11x1000; frotis de sangre periferica: Linfocitosis la mayoría de aspecto maduro, ocasionalmente con citoplasma amplio, levemente basófilo, y prolongaciones citoplásmicas. Cuantificacion inmunoglobulinas: IgG 1310, IgA 151, IgM 28, IgE 6.
ASPIRADO MEDULA OSEA: Abundantes megacariocitos en diferentes estadios madurativos. Serie roja hiperplasiada en cuantía moderada sin diseritropoyesis. Serie blanca con moderada degranulación en las formas más maduras aunque con presencia de todos sus elementos. Eosinofília del 7%. Además mostró una infiltración linfoide polimorfa que representaba el 31% de la celularidad, constituida por linfocitos de aspecto normal y otros de mayor tamańo, con citoplasma amplio y basofílico, sin nucleolo. El 9% de los linfocitos presentaba un citoplasma desflecado.
MARCADORES INMUNOLOGICOS EN SANGRE PERIFERICA: CD 19(+), CD 22(+), CD 23(-), FMC 7 positivos (37%), Kappa positivos, Lambda(-), CD 5: 19% de linfocitos T residuales, CD 103 positivo 35%, CD 25(-), CD11c positivo 86%, CD 10(-). ESTUDIO ADN: Coeficiente de variación 3.32, Fase Go/G1:99.8, Fase S 0.1, Fase G2/M 0.1. Índice ADN no aneuploidía.
CITOQUIMICA EN SANGRE PERIFERICA: Fosfatasa ácida: positiva difusa débil en todos los linfocitos. Inhibición L. Tartárico: No existe tartrato resistencia.
TAC TORACO-ABDOMINAL: Adenopatías mediastínicas e hiliares izquierdas. Derrame pleural derecho que produce atelectasia del lóbulo inferior. Hepatomegalia homogénea sin LOEs (Lesión Ocupante de Espacio). Esplenomegalia gigante que tras inyección demuestra varios defectos de repleción. Compresión y desplazamiento gástrico a la derecha. Adenopatías en ligamento gastrohepático y retroperitoneales. Útero de aspecto involutivo con una zona hipodensa en centro de la cavidad uterina. Rińón izquierdo comprimido y desplazado hacia atrás que conserva su funcionalidad.
BIOPSIAS PLEURALES y HEPATICAS: Compatibles con infiltración por un proceso linfoproliferativo de células uniformes de talla media con sus núcleo en disposición central y nucleolo prominente. BIOPSIA DE MEDULA OSEA: Descrita como cambios reactivos inespecíficos sin fibrosis.
El fenotipo inmunológico de este proceso linfoproliferativo se estableció como compatible con una Tricoleucemia variante de células B con restricción isotípica kappa.
HALLAZGOS HISTOLÓGICOS Y ULTRAESTUCTURALES.
Se hizo biopsia de la lesión cutánea referida e histológicamente nos encontramos con una densa celularidad compuesta por elementos isomorfos de talla media y de aspecto reniforme, que insinuaban en algunos campos una distribución nodular no bien definida. Se pudo distinguir en éstas células un citoplasma moderadamente abundante y que ocasionalmente adoptan la apariencia en "huevo frito" con un núcleo central y citoplasma claro alrededor. Éstas células era de talla media con sus núcleo en disposición central y nucleolo prominente. No había infiltración de la epidermis (figuras 2 y 3). En las imágenes mostradas podría interpretarse la presencia de un pseudosinusoide formado por un pequeńo lago de células rojas rodeadas de célula monocitoides, y una imagen central difícil de interpretar pudiendo sospecharse un fenómeno de emperipolesis como se ha sugerido en la discusión del caso. Esta imagen es desconocida fuera de las localizaciones mencionadas.
En la imagen ultraestructural (figura 4) observamos que las células tenían las prolongaciones citoplasmáticas características tal como se describen en los tricoleucocitos (que se encuentran en toda la periferia de la circunferencia celular a diferencia de las que se muestran en los linfomas de la zona marginal esplénica o la de los linfocitos pilosos circulantes) pero hay ausencia de cuerpos lamelares que suelen ser evidentes y numerosos en la tricoleucemia común y descritos como ausentes en la Variante Tricoleucémica.
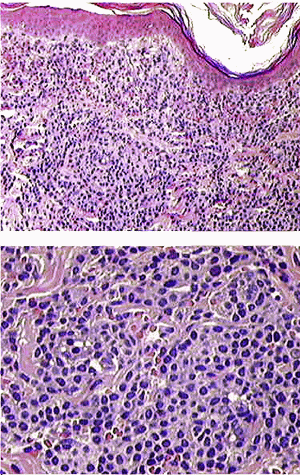
Figura 2
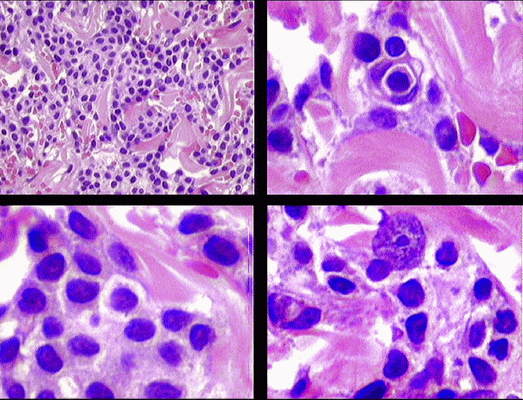
Figura 3
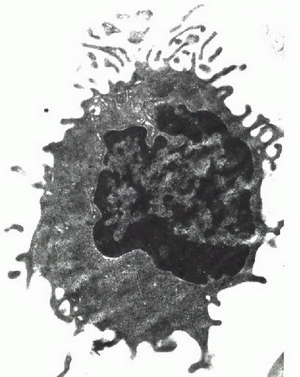
Figura 4
EVOLUCIÓN CLÍNICA DE LA PACIENTE
Instaurado el tratamiento con Interferón alfa 2b regresaron las lesiones cutáneas en su totalidad a las tres semanas pero persistió el derrame pleural y se incremento la visceromegalia. Ante estos datos de progresión se inició un tratamiento con Pentostatina durante 15 días no obteniéndose ninguna respuesta. La paciente falleció con enfermedad activa a los 5 meses del diagnóstico.
DISCUSIÓN
Las manifestaciones cutáneas de las leucemias se pueden clasificar en específicas (leukemia cutis) e inespecíficas. Las lesiones específicas se describen habitualmente como pápulas hemorrágicas, nódulos o placas de diverso tamańo así como lesiones violáceas o marrón rojizas como era nuestro caso. Los lugares frecuentes donde se pueden presentar son en la cara, extremidades o en el tronco.
Aunque las lesiones específicas en las LCP pueden presentarse en un 8% de los casos, no resultan tan infrecuentes como generalmente se asume aunque si es raro que una LCP debute con lesiones cutáneas específicas1. Ésta circunstancia se conoce que es más frecuente en los casos de los desórdenes linfoproliferativos de células T que en las leucemias de células B.
Las lesiones cutáneas inespecíficas son más frecuentes que las específicas pudiendo verse en más de un 40% de los pacientes con leucemia. Petequias, púrpura, equimosis, micosis y viriasis cutáneas, ectima gangrenoso, herpes zoster, pioderma grangrenosa, síndrome de Sweet, eritrodermia, ampollas intracutáneas, eritemas multiforme, urticaria, hiperpigmentaciones, xantomas y prurito son algunas de las lesiones inespecíficas descritas comúnmente en pacientes con leucemias. De entre éstas, la urticaria crónica es una de las lesiones cutáneas inespecíficas que pueden presentarse como signo en el debut de una LCP.
Finan y col. 2revisaron la prevalencia de hallazgos cutáneos en 113 casos de LCP encontrado que tan solo uno había presentado lesiones cutáneas específicas. De toda la bibliografía consultada 3-8 y hasta donde hemos podido conocer, nuestro caso es la primera LCP Variante descrita con lesiones específicas cutáneas.
El avance en el tratamiento con Interferón alpha es una modalidad terapéutica importante en el manejo de pacientes considerándose como la primera opción en aquellos pacientes que no pueden recibir transplante de médula ósea. Se han descrito respuestas clínicas favorables de pacientes con LCP a los que se ha instaurado un tratamiento mediante interferón alfa que superan el 90% de los ensayos clínicos practicados lo que hace que llevado a considerarlo como una medicación de primera línea para el tratamiento la leucemia de células peludas9.
Nuestro caso debe considerarse como una enfermedad progresiva en estado avanzado dada su visceromegalia, la infiltración medular y la presencia de adenopatías tanto mediastínicas como retroperitoneales por lo que el pronóstico a corto plazo era pobre. Además a la forma variante de éste proceso se le reconoce también una mala evolución con peor respuesta al tratamiento que la forma clásica. Los casos habituales de LCP suelen presentar tricoleucocitos típicos en sangre periférica y despertar su sospecha ante toda pancitopenia. En los casos en los que no es posible encontrar tricoleucocitos en sangre periférica, el estudio de la médula ósea se hace obligado. Cuando esto no es suficiente para hacer el diagnóstico final se aconseja la esplenectomía y el estudio histopatológico del bazo que suele encontrarse infiltrado por células características en su pulpa roja.
BIBLIOGRAFIA.
1.- Eiichi A, Shigeo I, Susumu I, Isao K. Specific skin lesions as the presenting symptom of hairy cell leukemia. Am J Clin Pathol. 1988 90(4):459-64.
2.- Finan, MC, Su WP, Li CY. Cutaneous findings in hairy cell leukemia. Journal of the American Academy of Dermatology 1984 11(5 Pt 1):788-97.
3.- Paoletti M, Bitter MA, Vardiman JW. Hairy-cell leukemia: morphologic, cytochemical, and immunologic features. Clin Lab Med 1988 8:179-195.
4.- Lazzaro B, Munger R, Flick J, Moriber-Katz S. Visualization of the ribosome lamella complex in plastic embedded biopsy specimens as an aid to diagnosis of hairy cell leukemia. Arch Pathol 1991 115:1259-1261.
5.- Cordone I, Annino L, Masi S, Pescarmona E, Rahimi S, Ferrari A, Giubilei E, Pignoloni P, Faraggiana T, Mandelli F. Diagnostic relevance of peripheral blood immunocytochemistry in hairy cell leukaemia. J Clin Pathol 1995 Oct, 48(10): 955-60.
6.- Dunn P, Shih LY, Ho YS, Tien HF. Hairy cell leukemia variant. Acta Haematol 1995 94 (2):105-8.
7.- Goyal R, Bajpai S, Chopra HK, Shinde SC, Badrinath Y, Sapre RS, Nair CN, Advani SH. Hairy cell leukemia--an unusual presentation. Leuk Res 1995 Jul, 19(7): 485-7.
8.- Clore LS Jr, Stafford CT. Chronic urticaria as a presenting sign of hairy cell leukemia. Allergy and Asthma Proceedings 1999. 20(1):51-5.
9.- Tokumine Y, Machii T, Inoue R, Shibayama H, Nishimori Y, Nakamura Y, Nojima J, Tagawa S, Kitani T. Effects of interferon-alpha on L-BCGF- and TNF-alpha-induced proliferation of hairy cell leukemia in Japan. Leukemia 1995 Jan, 9(1):25-9.
Comentario del Revisor Dr. Oscar Marín . Hospital Pablo Soria de Jujuy. San Salvador de Jujuy. Argentina
Leucemia de Células Pilosas es una neoplasia de pequeńas células linfoides, de estirpe celular B, infrecuente. Los autores nos presentan un interesante caso, de la rara Leucemia de Células Pilosas "Variante"; con una inusual forma de presentación como afección cutánea primaria. Los casos de estas variantes presentan rasgos histológicos similares a la forma clásica, pero las células circulantes presentan voluminosos nucleolos que recuerdan a prolinfocitos y tienen inmunofenotipo B; pero a diferencia de la forma clásica no expresan CD25. Entre los diagnósticos diferenciales se incluyen el Linfoma Marginal Esplénico con linfocitos vellosos circulantes y la Leucemia Prolinfocitica Crónica.
El caso que nos es presentado aquí tiene una forma de presentación considerada infrecuente como es la localización primaria, en sitio de afectación cutánea. El cuadro histológico observado, las características de las células circulantes y la expresión inmuno-histoquímica caracterizan fuertemente el diagnóstico realizado. La evolución agresiva es acorde a lo descrito en estos casos. Se hace una detallada descripción clínica y de los hallazgos hematológicos e histológicos. Un interesante e infrecuente caso.
Comentario del Revisor Dr. Bayardo Flores. Istituto Cantonale di Patologia. Locarno. Suiza
El Dr. Moro-Rodriguez y colaboradores nos presentan un caso de una entidad hematológica extremadamente infrecuente y que requiere un exhaustivo estudio para poder establecer el diagnóstico y descartar una serie de posibles diagnósticos diferenciales.
Estoy seguro que el esfuerzo de los autores será bien apreciado por los amantes de la hematopatología ya que el caso en cuestión además de su rareza presenta una particularidad clínica no informada en estudios precedentes como es su presentación inicial con manifestaciones cutáneas específicas (leucemia cutis).
La leucemia de células peludas es un proceso clonal de células B con aspectos clínicos, morfológicos e inmunofenotípicos particulares, descrita inicialmente como una retículoendoteliosis leucémica afecta predominantemente a hombres de media edad y usualmente se presenta con pancitopenia, infecciones, esplenomegalia, un número variable de células peludas circulantes y una médula ósea con alto grado de fibrosis. Inmunofenotípicamente las células malignas expresan restricción de cadenas ligeras y positividad a CD19, CD20, CD22,,CD11c, CD25, FMC7 y CD103 y negatividad para CD5, CD10 y CD23. El tratamiento con interferón- alfa, deoxicoformicina, cladribina y la esplenectomía, en general resultan efectivos con remisiones completas hasta de un 90%.
En 1980 Cawley y cols. describieron la entidad que se conoce como Tricoleucemia "Variante", un síndrome linfoproliferativo extremadamente raro, cuya ocurrencia se ha estimado en sólo 1.1% en comparación a la forma clásica de tricoleucemia y difiere de ésta, tal como lo establecen los autores de este estudio, en diversos aspectos; pacientes de mayor edad, linfocitosis y ausencia de infeccio-nes. Inmunofenotípicamente las células de la forma variante se distinguen de las de la forma clásica principalmente por la negatividad con el CD25, variable expresión del CD103 y mayor expresión de CD11c.
La evolución clínica de la Variante Tricoleucémica se caracteriza por mayor agresividad, poca o ninguna respuesta a los tratamientos convencionales de la forma clásica y un mal pronóstico.
El Dr. Moro-Rodriguez y cols. agotaron los medios diagnósticos posibles cumpliendo con los criterios necesarios para clasificar el caso como una verdadera Tricoleucemia "Variante", que además se presentó con un cuadro clínico del todo particular, con manifestaciones cutáneas que orientaban mas bien hacia otros posibles diagnósticos lo que lo hace todavía más aleccionador.
Comentario del Revisor Prof. Reynaldo Falcón-Escobedo Profesor de Anatomía Patológica, Facultad de Medicina, Universidad Autónoma de San Luis Potosí, San Luis Potosí, S.L.P. México
El artículo vale la pena, es una presentación inusual y aparentemente la primera en la literatura con afección dérmica como manifestación inicial.
Poco que agregar a los brillantes comentarios de los Dres. Oscar Marín y Bayardo Flores.
La OMS define la Leucemia de células vellosas (Tricoleucemia) como una neoplasia de células linfoides B pequeńas, con núcleos ovales y abundante citoplasma con proyecciones "vellosas" en médula ósea y sangre periférica que infiltran difusamente médula ósea y la pulpa roja esplénica. Estas células expresan fuertemente CD103, CD22 y CD11c. La variante de esta entidad es una enfermedad muy infrecuente caracterizada por una histología en médula ósea y en bazo semejantes a la Tricoleucemia típica, pero con células circulantes con núcleo oval o redondo y un nucléolo prominente (que semejan prolinfocitos) y citoplasma velloso moderadamente basófilo. Típicamente los pacientes presentan leucocitosis sin monocitopenia. Las células muestran inmunofenotipo B con expresión de IgG membranal, sin expresión de CD25.
El caso presentado por el Dr. Ernesto Moro-Rodríguez y colaboradores está perfectamente documentado clínicamente y por laboratorio, así como bellamente ilustrados histológica y ultraestructuralmente, por lo que el diagnóstico es inobjetable.
Las fotomicrografías no. 2 muestran en efecto un aspecto vagamente nodular y citomorfología monocitoide que recuerdan a un linfoma de la zona marginal, que en una biopsia de piel sin mayores datos clínicos puede plantear serios problemas de diagnóstico diferencial. Aunque la microscopía electrónica no deja lugar a dudas, hubiera sido deseable conocer los hallazgos de inmunohistoquímica en la biopsia de piel.
El caso nos deja una gran enseńanza tanto por la acuciosidad diagnóstica de los autores (ˇfelicitaciones!), como por la posibilidad de afección cutánea en esta entidad.
Enhorabuena a los lectores del artículo del Dr. Moro y cols. que seguramente lo disfrutarán tanto como los comentaristas y editores de la Revista Electrónica de Biomedicina.
Recibido: 22 de mayo de 2004. Recibido revisado 29 de Agosto de 2004.
Publicado: 14 de Septiembre de 2004
Correspondencia:
Prof. Dr. Ernesto Moro Rodríguez
Universidad Rey Juan Carlos
Facultad de Ciencias de la Salud
Av de Atenas s/n
28922 Alcorcón (Madrid)