Indice del volumen Volume index
Comité Editorial Editorial Board
Comité Científico Scientific Committee
LINFOMA T DE TIPO ANGIOINMUNOBLASTICO:
Estudio clínico-patológico del Grupo de Estudio de Linfomas de Jujuy (GELJ)
Oscar Marín1 Viviana Gloria Hope2, Gabriela Lamas Hernández3,
Rebeca Mérida3, Eugenia Fandińo4
Ana Paula Gaite4, Liliana Quispe4, Graciela Berlingieri5, Ana Carolina Ituarte5, Ana Laura Reynaud6.
1Servicio de Anatomía Patológica, 4Residencia de Clínica Médica y 5 Servicio de Oncología, Hospital "Pablo Soria".
2Clínica Médica, Sanatorio Quintar. 3Laboratorio de Bioquímica CAMING.
6 Laboratorio Privado de Patología y Citopatología.
Integrantes del Grupo de Estudio de Linfomas de Jujuy (GELJ). San Salvador de Jujuy. Argentina
Rev Electron Biomed / Electron J Biomed 2005;1:55-63
- 1) Borramiento total de la histoarquitectura con infiltración de la cápsula y tejido circundante. Ausencia de centros germinales.
- 2) Citología Polimorfa, de pequeńo, mediano y gran diámetro, con un porcentaje de células claras.
- 3) Presencia de células reactivas como plasmocitos, granulocitos eosinófilos, histiocitos y células epitelioides.
- 4) Marcado incremento de vénulas epitelioides, arborescentes con engrosamiento PAS positivo de la membrana basal.
- 5) Nidos o mantos de Células Reticulares Dendríticas (DRC).
- 6) inmunofenotipo de células T. Cuando al menos 5 de estos criterios fueron satisfechos los casos se incluyeron en este trabajo.
Comentario del revisor, Dr. Antonio Felix Conde Martín. Hospital Infanta Cristina. Badajoz. Espańa.
Comentario del revisor, Dr. Rodrigo Valdés Annunziata. Laboratorio de Patología Histonor. Antofagasta. Chile
Comentario del revisor, Dr. Hernan Molina Kirsch Laboratorio de Patología. Ciudad de Guatemala. Guatemala.
ABSTRACT
Jujuy have a high incidence of lymphomas and among these a high number of T-cell lymphomas is observed. However the Angioimmunoblastic T-cell Lymphoma once a best characterized T-cell lymphomas, was not studied in Jujuy in both, incidence and clinicopathologic features. These lymphoma present a characteristic clinical findings and a distinctive histopathological picture with T-cell immunophenotype that permit a clear differentiation from other peripheral T-cell lymphomas.
Angioimmunoblastic Peripheral T-cell lymphoma represent the 9,1% of our cases of lymphomas, registered between 1985 and July 2004, when we consider nodal and extranodal cases, but represent 18% among the nodal lymphomas. All the cases studied presenting the distinctive histopathologic picture, with effacement of the lymph node architecture, absence of germinal centers, polymorphic cytology, marked increase of arborizing vessel PAS-positive and accumulations of Follicular Dendritic Reticular Cells, CD21+. Clinically we observed generalized lymphadenopaty, skin lesions, pulmonary manifestations, systemic symptoms and aggressive behavior.
Keywords: T-cell lymphoma, Angioimmunoblastic T-cell lymphoma, non-Hodgkin's lymphoma, Peripheral T-cell Lymphomas.
RESUMEN:
Jujuy es una zona de alta incidencia de linfomas y entre ellos se registra un elevado número de linfomas de células T. Pese a esto el linfoma T tipo linfadenopatía Angioinmunoblastica, uno de los mejor caracterizados linfomas de células T, no ha sido estudiado, tanto en su incidencia, como en sus caracteres clínico-patológicos. Estos linfomas presentan un cuadro clínico característico y un distintivo cuadro histopatológico e inmunofenotípico, que permiten su diferenciación de otros tipos de linfomas T.
Este linfoma representa el 9,1% de los casos registrados en nuestro hospital entre 1985 y julio de 2004, cuando se consideran casos ganglionares y extraganglionres y el 18% de los casos ganglionares. Presentan el característico cuadro histopatológico con borramiento de la histoarquitectura ganglionar linfática, citología polimorfa, marcado incremento de vénulas postcapilares y mantos de células reticulares dendríticas positivas para CD21. Clínicamente se observó poliadenopatía generalizada, lesiones cutáneas, manifestaciones pulmonares, fenómenos alérgicos, síntomas sistémicos, estadio avanzado de enfermedad y una agresiva evolución clínica
Palabras clave: Linfoma de células T, linfoma T angioimmunoblástico, Linfoma no Hodgkin, Linfoma T periférico.
INTRODUCCION:
La provincia de Jujuy; localizada en el noroeste de Argentina, es una zona de alta incidencia de linfomas y entre ellos a diferencia de lo reportado para países occidentales, un importante número de linfomas T es observado. Pese a ello; el linfoma T tipo linfadenopatía Angioinmunoblastica -uno de los mejor caracterizados linfomas de células T- no ha sido estudiado, tanto en sus caracteres clínico-patológicos, ni en su incidencia en nuestra provincia. Quizás opacado por la presencia de linfomas T considerados infrecuentes en países occidentales como Linfomas T relacionados a HTLV-1 (Human T-cell Lymphoma/Leucemia Virus Type-1), denominados ATLL (Adult T-cell lymphoma/Leukaemia) y linfomas de células T/Natural Killer que han focalizado nuestra atención.1, 2.
El linfoma de tipo Linfadenopatía Angioinmunoblastica (AILT) es una afección linfoproliferativa neoplásica maligna, de células linfoides T periféricas, caracterizada por enfermedad sistémica, afectación predominantemente ganglionar con prominente proliferación de vénulas epitelioides y de células foliculares dendríticas3. Ocurre en pacientes de edad media o en adultos ańosos, con igual incidencia de sexos y usualmente con linfadenopatía generalizada, hepatoesplenomegalia y frecuente rush cutáneo3, 4. En general los pacientes presentan estadio avanzado de enfermedad, con síntomas sistémicos, prurito e hipergammaglobulinemia policlonal. Otros síntomas descritos son edema, efusión pleural, artritis y ascitis3, 4.
En 1971 en un Workshop en Nagoya, Dorfman describió una entidad distintiva, observada en 4 pacientes, publicado luego por Liao y col5. Otros autores participando en el mismo Workshop reportaron luego similares lesiones bajo diferentes nombres. Lennert la denominó Linfogranulomatosis X (LgrX)6, mientras que Lukes y Tindle la denominaron como Linfadenopatía Inmunoblastica (IBL)7 y finalmente Frizzera y col. propusieron el término Linfadenopatía Angioinmunoblastica con disproteínemia (AILD)8. Todos estos autores describieron está afección como un proceso reactivo atípico de naturaleza autoinmune. Transformación en incuestionables casos de linfoma inmunoblastico, fue reportada en varias publicaciones en un porcentaje de casos de AILD y entonces esta entidad comenzó a considerarse como una afección probablemente preneoplasica9.
En 1979, Shimoyana y col. en Tokio describieron una lesión similar, pero la interpretaron esta vez como un linfoma y denominándolo Linfoma T tipo-Linfadenopatía Angioinmunoblastica (IBL-Like T-cell Lymphoma)10, debido a la similitud con el proceso denominado IBL y desde entonces reconocido como un linfoma, con abundante descripción en la literatura dentro y fuera de Japón, reportando su naturaleza clonal11-17.
Actualmente mas del 90% de casos de AILD han demostrado ser proliferaciones T monoclonales y es ahora uno de los linfomas T mejor caracterizados, integrando los linfomas a predominio ganglionar linfático (node-based lymphomas), junto al linfoma de Grandes Células Anaplásicas (ALCL) y a los linfomas de Lennert y Linfoma de la Zona T, ambos incluidos ahora dentro de los Linfomas T periféricos de tipo no especificado (PTCL-NOS), en la nueva clasificación de la OMS18 y previamente reconocido como tales en las clasificación de Kiel (1975)19 y también incluídos como PTCL-NOS en la clasificación de REAL (Revised European American Lymphoma classification)(1994)20.
Se reporta en la literatura la transformación de un porcentaje de Linfoma T tipo AILT, en linfomas agresivos de células B a partir de oligoclones de células B presentes en estos linfomas y que pueden dar lugar a la formación de monoclones de células B implicándose al virus de Epstein-Bar en estos casos21, 22.
En nuestro servicio se encontraban 7 casos con diagnóstico de linfoma T tipo AILT identificados luego de un trabajo de reclasificación de toda la patología linfoide de nuestro hospital, tarea que demandó 10 ańos. Opacados por la presencia de casos de ATLL y de linfomas Natural Killer, no habíamos profundizado los estudios sobre este linfoma. La llegada de 4 nuevos casos consecutivos de linfomas T AILT, motivó la formación de un grupo interdisciplinario para conocer las características clínico-patológicas de estos linfomas en nuestro medio. Si bien 7 de estos casos ya tenían diagnostico de linfoma AILT, solo habían sido estudiados en su anatomía patológica, desconociéndose la clínica y datos de laboratorio de estos pacientes y su evolución. Por lo tanto se centro la atención de estos casos hacia aspectos anatomo-clínicos. La intención de este trabajo entonces es analizar la incidencia y las características clínico-patológicas de casos de Linfoma T tipo AILT registrados en el hospital cabecera de la provincia de Jujuy. Y primordialmente conocer si la evolución de estos era la de un linfoma agresivo o un proceso hiperinmune. Para ello se creó un grupo multidisciplinario constituido por anatomopatólogos, bioquímicos, médicos oncólogos y médicos clínicos, quienes se encargaron de revisar el material histológico e inmunohistoquímico, la incidencia, los datos de laboratorio, las características clínicas de este grupo de pacientes y su respuesta a la terapéutica y curso clínico
MATERIAL Y MÉTODOS
Entre julio de 1985 y julio de 2004 se registran en el Servicio de Anatomía Patológica del Hospital "Pablo Soria" de Jujuy, 428 casos de linfomas diagnosticados de acuerdo a la clasificación de la OMS para neoplasias hemolinfoides (2001) (World Health Classification of Tumours, Pathology and Genetics, Tumours of Haematopoietic and Lymphoid Tissue, IARC press 2001) La distribución porcentual de estos resultó: 62 (15%) casos de linfomas de Hodgkin (Enfermedad de Hodgkin) y 366 casos de linfomas no-Hodgkin, incluyendo 123 (28%) casos de linfomas T y 243 (56%) casos de linfomas B. Entre los linfomas T se encuentran 11 casos, con diagnóstico de linfomas T de tipo AILT, que son la base de este estudio. Se tomo además del archivo un caso para su comparación, con similares características histológicas en un paciente pediátrico, que procedía como ínterconsulta, del medio privado con diagnóstico de linfoma, no incluyéndose en nuestra serie como linfoma. Entre los casos de linfoma; uno procedía también como ínterconsulta del ámbito privado y los restantes pertenecían al hospital Pablo Soria.
Para este linfoma utilizamos la definición de la OMS quien lo define como un linfoma de células T periféricas, caracterizado por enfermedad sistémica, infiltrado polimorfo, involucrando ganglio linfático, con prominente proliferación de vénulas de endotelio alto y células foliculares dendríticas.
Los criterios histológicos utilizados en este trabajo para ser considerados casos de linfomas AILT, tomados de Lennert19 son los siguientes:
Se realizó estudio inmunohistoquímico en los primeros 7 casos durante una investigación de casos de ATLL. Se neutralizó la peroxidasa endógena por incubación de las secciones en H202 al 3% en metanol por 30 minutos. Recuperación antigénica fue realizada por incubación de secciones en 0,01 mol/L buffer citrato a ph 6.0, seguido por 5 minutos en autoclave. Las secciones han sido expuestas a los anticuerpos primarios por 2 horas, utilizando los siguientes: CD3 (Dako®-CD3, A0452), CD4 (NCL-CD4-IF6-novocastra), CD8 (CD8 L8/144B Dako®, Glostrup, Denmark), CD20 (CD20cy L26-Dako®), CD21 (clone 1F8-Dako®) y CD30 (NCL-CD30-clon Ber-H2; Dako®). En algunos casos se estudiaron además las cadenas livianas de inmunoglobulinas Kappa y Lambda (Immunotech®; Marseilles-France). La unión con anticuerpos primarios fue visualizada por Elite ABC Method (Vector laboratory®, Burlingame, CA, USA) y reacción DAB-H202. Luego de contratinción nuclear, se deshidrataron a través de series de etanol graduado y lavados en xileno. Las secciones fueron montadas en medio plástico. Este proceso fue realizado en la Universidad de Kagoshima, Japón realizados sobre un amplio grupo de linfomas T algunos de los cuales tenían diagnostico previo de AILT y pertenecían a casos de ATLL, habiendo quedado en archivo, ya que los estudios se orientaban hacia otro tipo de linfomas. Los restantes 4 casos fueron enviados la Fundación para le Medicina de la ciudad de Córdoba-Argentina, donde se realizó inmunohistoquímica mediante sistema de tinción automática (Autostainer DAKO), con anticuerpos primarios DAKO, al carecer nuestro servicio de inmunohistoquímica actualmente se terceriza este servicio en Córdoba.
Se revisaron los datos clínicos y de laboratorio contenidos en la historia clínica, orientados a buscar datos relacionados a edema, prurito, fármacodermia, manifestaciones pulmonares, anemia, hipergammaglobulinemia y fenómenos alérgicos. Además de pérdida de peso, anorexia, astenia y fiebre. Así también la evolución clínica de los mismos.
RESULTADOS
a) Hallazgos Clínicos y de Laboratorio:
El rango de edad de los pacientes fue de 23 a 64 (media de 41), salvo un caso masculino de 23 ańos de edad el resto de los casos con edad superior a 50 ańos. Un caso pediátrico con cuadro histológico similar, examinado para su comparación tenía 10 ańos.
Los pacientes presentaron poliadenopatías en todos los casos, fiebre, pérdida de peso, anorexia, astenia y prurito. Otros hallazgos fueron los siguientes: hepatomegalia, esplenomegalia, alteraciones mediastinales (hilio aumentado de volumen), tos crónica, micosis superficiales, muget oral, y edema de miembros inferiores
Se encontró además presencia de exantemas cutáneos, prurigo y eccemas. también se registra un caso con fármacodermia. Los datos clínicos se resumen en Tabla 1.
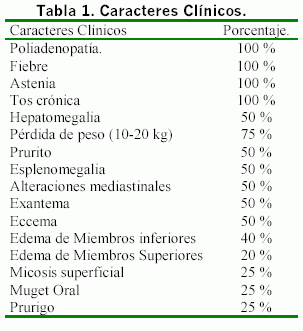
Las adenopatías más frecuentes fueron: inguinales, axilares, cervicales, supra-clavículares y sub-mandíbulares. La ubicación topográfica de estas se resume en Tabla 2.

Entre los hallazgos de laboratorio se encuentran anemia e hipogammaglobulinemia severa como factores destacados, estudiándose en algunos casos HIV y HTLV-1 que fueron negativos en todos ellos. LDH se presentó elevada en todos los pacientes estudiados.
Se encontraron alteraciones de laboratorio positiva para Tuberculosis y VDRL positiva en un caso cada uno respectivamente. Los datos de laboratorio se resumen en la tabla 3.

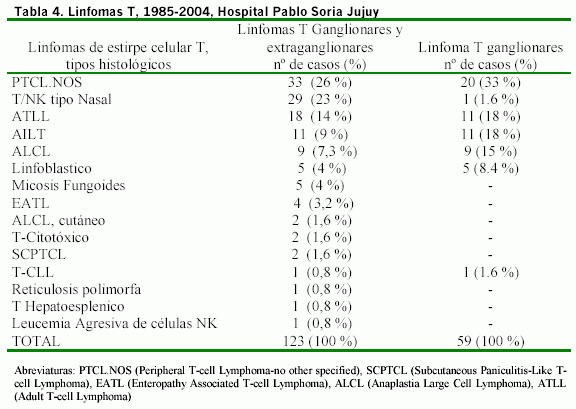
Mientras que la incidencia de estos linfomas entre nuestros casos de linfomas T es comparada incluyendo casos ganglionares y extraganglionares, en tabla 4.
b) Hallazgos Histológicos:
Se observó en todos los casos, el característico cuadro histológico con reemplazo de la histoarquitectura ganglionar linfática por proceso proliferativo, con patrón histológico difuso formado por linfocitos de pequeńo y mediano diámetro, con caracteres nucleares poco agresivos, con núcleos levemente irregulares y nucleolos inconspicuos. Y la característica presencia de prominentes vasos sanguíneos arborescentes de endotelio alto, reconocidos como vénulas post-capilares, mejor observados con técnicas que PAS (Fig.1) y cuyas membranas basales son positivas con esta coloración.
En algunas áreas se observan nidos de células claras (fig.2) y es característico el frecuente hallazgo de grupos celulares, de células fusiformes eosinofílicas que en ocasiones se presentan como nidos celulares y menos frecuentemente en algunos casos, presentan caracteres de folículos linfoides de tipo "Burned-out".
Estos grupos celulares han sido identificados como células reticulares dendríticas, mediante inmunomarcación para CD21 (Fig.3), siendo un hallazgo característico y constante en estos linfomas. La mencionadas células reticulares dendríticas (DRC) se expanden en el estroma, por fuera de folículos linfoides, característicamente a partir de un vaso sanguíneo.
En alguno de estos casos se observaron células de mayor diámetro y morfología similar a células de Reed-Sternberg. Un hallazgo frecuente es el de células gigantes de tipo cuerpo extrańo (Fig. 4).
 Figura 1. Vénulas epitelioides PAS+ |
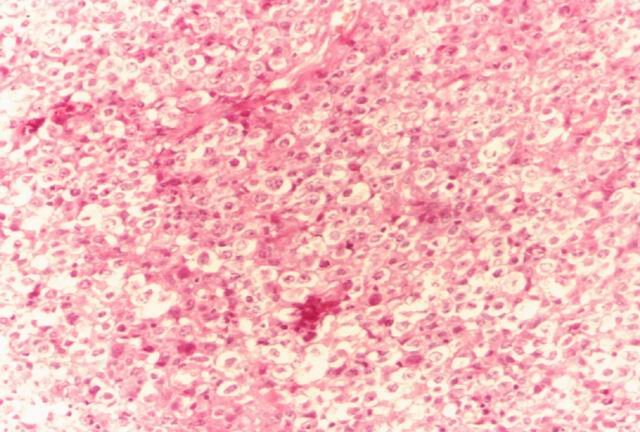 Figura 2. Células claras (PAS). |
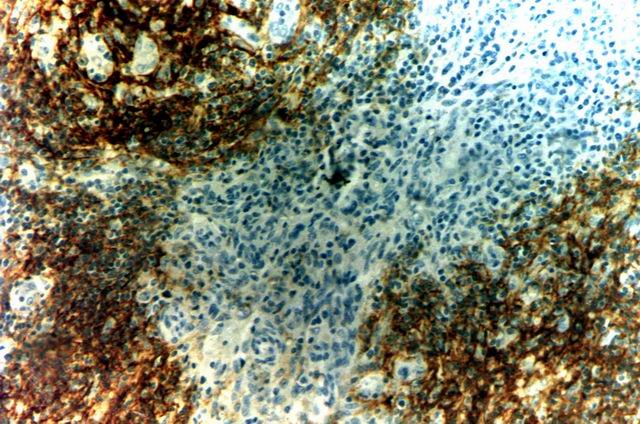 Figura 3. Células Reticulares Dendríticas CD21+ |
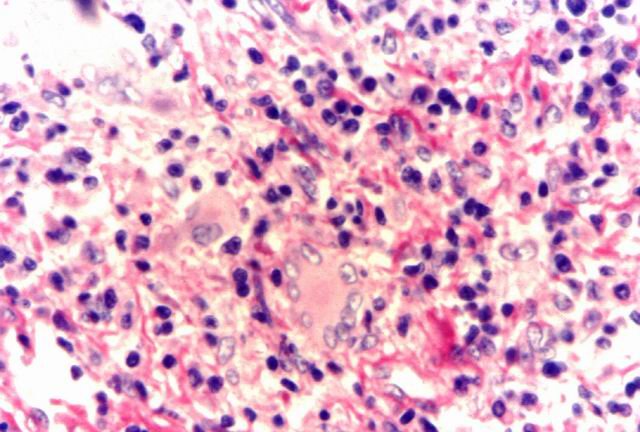 Figura 4. Célula gigante tipo cuerpo extrańo. |
El estudio inmunohistoquímico demostró que las pequeńas y medianas células son positivas para CD3 (Fig. 5) y CD4, mientras que células de mayor diámetro son reconocidas como CD8 (Fig. 6) positivas, identificables como linfocitos de estirpe celular T, con una mezcla de tipos celulares "helper" y "supresor". En todos los casos se identificaron además grupos celulares positivos para CD20, a manera de nódulos (Fig. 7), coincidiendo en general con áreas de células reticulares dendríticas (DRC) marcadas con CD21 y pertenecientes entonces a células linfoides de linaje B.
Uno de los casos registrados en nuestro archivo y tomado para comparación, corresponde a un paciente pediátrico de diez ańos de edad, y que presenta sin constituir un linfoma, similares caracteres histológicos a los mencionados previamente con la excepción, de la presencia de folículos linfoides reactivos con centros germinales en actividad, y constituyendo el estadio de hiperplasia folicular reactivas en fase II, con su característico aspecto del cielo estrellado y presencia de corona de manto linfocitario. Su evolución fue buena sin tratamiento.
Solo un caso de linfoma presenta caracteres histológicos diferentes a los descrítos; habiéndose obtenido del mismo tres biopsias. La primera de ellas, realizada en 1987 presentaba, además del cuadro histopatológico ya descrito, un extraordinario número de granulocitos eosinófilos, tanto en la biopsia ganglionar como en el estudio de médula ósea. La segunda biopsia efectuada en 1990 presentaba un elevado número de células epitelioides (Fig.8), habiendo desaparecido los granulocitos eosinófilos. La tercer biopsia también realizada en 1992 presentaba el típico cuadro de un linfoma AILT. El caso fue tratado solamente con corticoides falleciendo poco después de la tercer biopsia. Excepto éste, los restantes casos de linfomas tipo AILT, presentaron el cuadro histológico ya descrito.
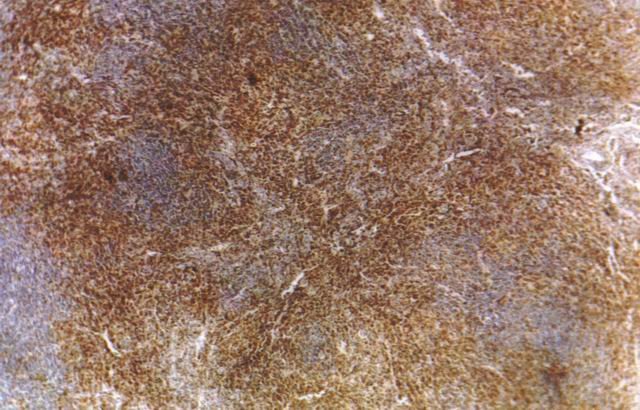 Figura 5. Linfocitos T marcados con CD3. |
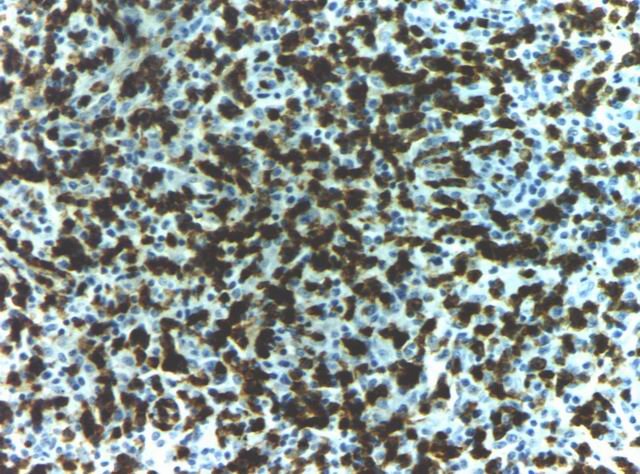 Figura 6. Células T, CD8+ (Supresoras) |
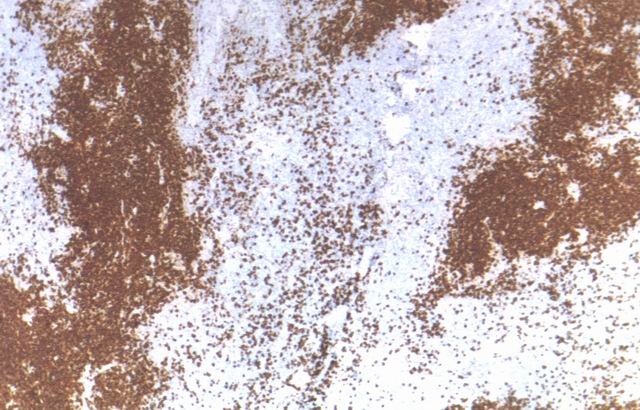 Figura 7. Nódulos de células B, CD20+ |
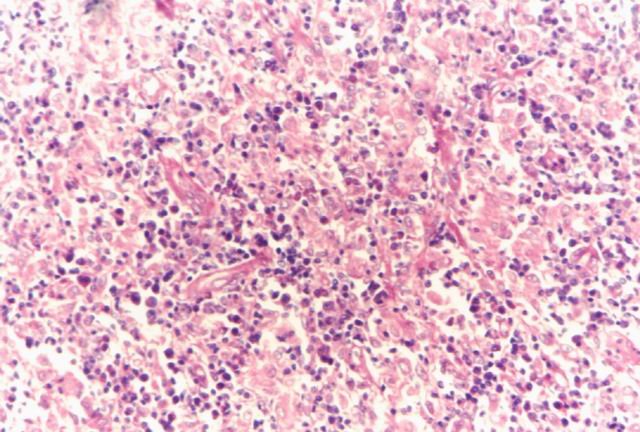 Figura 8. Células epitelioides. |
c) Incidencia:
Entre 123 casos de linfomas de células T al linfoma tipo AILT, le corresponde el 9% de los linfomas T, cuando se incluyen las formas ganglionares y las extraganglionares, sin embargo entre las formas ganglionares puras constituyen el 18%, superados por el linfoma PTCL-NOS e igualando los casos de ATLL y superando al linfoma tipo ALCL. La alta incidencia de linfomas extraganglionares (63%) entre nuestros casos de linfomas T, hace que dentro del espectro de linfomas observados en nuestro estudio, AILT no represente un gran porcentaje, pero es el segundo tipo de infoma T entre aquellos de ubicación ganglionar, superado solo por PTCL-NOS e igualando a los casos de ATLL. (Tabla 4). Es necesario aclarar que se trata de incidencia hospitalaria, pero al recibir nuestro hospital la casi totalidad de biopsias de la provincia, las cifras de este estudio en alguna manera, pueden dar una idea de la incidencia relativa de los linfomas en la provincia.
d) Distribución por sexos:
Si bien se reporta una distribución por sexos hombre: mujer de 1,2:1 en nuestros casos 10 de los pacientes (90%) han sido masculinos y un solo caso femenino, llama atención la gran desproporción entre sexos, siendo una de las mas marcadas diferencias entre los diferentes subtipos de linfomas T, en nuestro archivo.
f) Tratamiento:
El tratamiento quimioterápico instituido en 9 casos consistió en esquema CHOP, falleciendo de 8 de estos pacientes entre 3 y 4 meses luego del diagnóstico original y lográndose remisión completa en uno de ellos. Uno de los casos (ya mencionado) fue tratado solamente con corticoides y falleció cinco ańos después del diagnóstico original mientras uno de los casos falleció a los quince días del diagnóstico sin poder recibir tratamiento de quimioterapia.
DISCUSION
Si bien se considera en la literatura a este tipo de linfoma, como uno de los más frecuentes y mejor caracterizados entre los linfomas de células T, sólo representa en nuestra serie el 9 % de los casos de linfoma de células T, en el período de tiempo estudiado. Siendo superados por linfomas de células T de tipo periférico-NOS (sin otra especificación), por el linfoma de tipo nasal NK/T y Linfomas de tipo ATLL (Adult T-cell Lymphoma /Leucemia) asociados a virus de HTLV-1 (Human T-cell Lymphoma/leukemia Virus, Type-1). Sin embargo superando a otros linfomas representativos de células T como la Micosis Fungoides y el linfoma Linfoblastico T. Pero cuando se consideran solo los linfomas ganglionares de células T su porcentaje asciende al 18% superado solo por los casos de PTCL-NOS e igualando a los casos de ATLL. El alto número de casos extraganglionares y la distribución porcentual de subtipos es mas similar en nuestro caso a las series Asiáticas que occidentales23, parece indicar esto un patrón genético que condicione su incidencia, condicionando la presencia de estos linfomas en población amerindia autóctona con rasgos antropomorfos de tipo mongoloide1 siendo además la presencia de casos de ATLL y linfomas de células NK/T responsables de esta frecuencia relativa disminuida.
El linfoma tipo AILT tiene descriptas características clínico-patológicas que nos interesaron en su estudio. Entre ellas la presencia de fenómenos asociados de tipo hiperinmune o autoinmune, como ser presencia de hipergammaglobulinemia, rash cutáneo, exantemas y reacciones autoinmunes o alérgicas a diferentes fármacos entre ellos antibióticos y quimioterápicos, que deseábamos evaluar si eran presentados entre nuestros pacientes. Entre nuestros casos no pudo constatarse la presencia de hipergammaglobulinemia, debido a que en muchos casos esta no fue estudiada, siendo imposible de evaluar en este estudio dado el carácter retrospectivo del mismo, debido al fallecimiento de la mayoría de los pacientes. Sin embargo en los últimos tres casos esta fue estudia encontrándose en 2/3 pacientes, en forma inversa a lo esperado, una hipogammaglobulinemia severa. En la mayoría de los casos pudo constatarse la presencia de alteraciones cutáneas de tipo exantemas, prurigo, eccemas y en uno de los casos pudo documentarse la existencia de fármacodermia. La alergia a diversos fármacos fue documentada en tempranos artículos7,24, pero luego en la literatura este dato no es encontrado.
Se encontraron la mayoría de los signos y síntomas descritos en estos linfomas, como ser múltiples adenopatías, hepatomegalia, esplenomegalia, prurito, compromiso pulmonar, edema de miembros inferiores y lesiones cutáneas.
Un elevado porcentaje de pacientes presenta síntomas B. De los pacientes que presentaban compromiso pulmonar, con manifestaciones como tos crónica, expectoración e imágenes radiológicas anormales, sólo uno de ellos fue estudiado mediante biopsia pulmonar presentando infiltrado linfocitario compatible con afección linfomatosa.
Los hallazgos histológicos han sido remarcablemente constantes con presencia de linfocitos de diámetro pequeńo a mediano, con morfología ligeramente irregular y frecuente presencia de células con citoplasma claro. En dos de nuestros casos el número de células claras fue muy elevado. Un hallazgo constante fue el de grandes "mantos" o agregados proliferativos de células reticulares dendríticas marcadas positivamente mediante CD21.
En algunos de estos casos las células reticulares dendríticas son encontradas constituyendo folículos de tipo "Burned-out".
Muy constante y característico es el hallazgo de numerosas vénulas de endotelio alto, con marcado carácter arborescente reconocibles como una hiperplasia de vénulas post-capilares de tipo epitelioide, mejor observables con técnica de PAS. Los hallazgos morfológicos hacen de este linfoma uno de los más característicos en su evaluación histológica, permitiendo ser diferenciados de otros tipos de linfomas, ya en base al estudio morfológico. Sin embargo en casos "borderline" su diagnóstico puede ser dificultoso.
El estudio inmunohistoquímico también ha sido muy constante demostrando las células neoplásicas inmunofenotipo de células T, con una mezcla de linfocitos CD3, CD4 positivos y otros CD8 positivos, de mayor diámetro e identificables estos últimos como linfocitos T de tipo "supresor". Esta expresión resulta muy característica del linfoma AILT ya que los restantes linfomas T/NK son CD4+ CD8- o CD4- CD8+, pudiendo ser doble negativos, pero esta doble positividad es un elemento que los diferencia de otros linfomas T periféricos. En todos los casos se ha observado nódulos de células linfoides de estirpe celular B, caracterizadas por la expresión inmunohistoquímica de CD20 y por coincidir el área topográfica donde ellas se encuentran, con las regiones identificadas mediante CD21 y constituidas por células reticulares dendríticas, constituyendo probablemente folículos linfoides remanentes, que han perdido su estructura histológica normal. Se conoce que las células reticulares dendríticas son las encargadas de constituir el micro-ambiente ganglionar linfático donde viven las células de linaje B y por lo tanto parecen representar folículos linfoides en involución cuyo estadio final es el que se describe frecuentemente en la literatura como folículos "burned-out".
En sólo dos casos de este estudio el patrón histológico presenta variantes a lo descrito. En uno de ellos la estructura histológica es remarcablemente similar a la descrita con la excepción de la presencia de folículos linfoides bien constituidos, con centros germinales activos y presencia de corona de manto linfocitario. Se trata de un caso pediátrico de diez ańos de edad, que procedía de ínterconsulta, con diagnóstico de linfoma y cuya evolución sin tratamiento fue favorable, pareciendo representar una hiperplasia ganglionar atípica. Este caso fue tomado del archivo para su comparación histológica y resulta sorprendente la similitud con los casos de linfoma. El restante caso presenta tres estudios de biopsia, con diferentes patrones celulares predominando en el estudio histológico y caracterizados por un extraordinario número de granulocitos eosinófilos en la primera biopsia, que planteaba diagnóstico diferencial con Linfoma de Hodgkin. Un muy elevado número de células epitelioides en la segunda biopsia con diagnóstico diferencial con Linfoma de Lennert y Linfoma de Hodgkin. En la tercera biopsia recién se pudo observar, el cuadro morfológico característico de un linfoma de tipo AILT. Por lo tanto solo uno de los casos de linfomas presentó variación en su patrón histológico, pudiendo tratarse de estadios tempranos, antes de constituir el característico cuadro histológico.
En todos los casos estudiados a excepción de aquellos mencionados como poseedores de caracteres y histológicos diferentes, la presentación clínica fue con estadio avanzado y mala evolución clínica, que salvo en un caso culminó con la muerte de los pacientes en un período de tiempo de entre quince días y cinco meses a partir del diagnóstico original.
Interesante es el hecho de que estos linfomas hayan sido descritos como afecciones reactivas o fenómenos ganglionares hiperinmunes, cuando la evolución clínica de los pacientes, tanto en nuestros casos como lo que se describe en la literatura, es notoriamente agresiva. Estos linfomas presentan características clínicas e histopatológicas que pueden llevar a la confusión de estos linfomas, con fenómenos reactivos hiperinmunes con la consecuencia de terapéutica inapropiada.
Si bien la interpretación inicial de estos linfomas como un proceso reactivo ha sido largamente desacreditada en la literatura, algunos autores insisten en la posibilidad de la existencia de un número de casos, que pueden pertenecer realmente a procesos reactivos25. Otros autores consideran que estos linfomas, o algunos de estos pueden ser precedidos de la fenómenos pre-neoplásicos, con proliferaciones oligoclonales antes de constituir un verdadero linfoma. La presencia en nuestro archivo de un caso pediátrico con similar cuadro histológico a estos linfomas, con buena evolución clínica a pesar de no haber tenido tratamiento, sugiere que en pacientes pediátricos la posibilidad de fenómenos hiperinmunes puede ser cierta. Es necesario aclarar que este caso no esta en nuestro archivo diagnosticado como linfoma, sino que fue enviado en ínterconsulta como este, y tomado para comparación histológica, ya que presentaba un cuadro morfológico similar al observado en linfomas. Lo que indica la peligrosidad de realizar un diagnóstico de este tipo en nińos o en casos donde se desconoce la edad del paciente.
En adultos lesiones histológicas con características de un linfoma de tipo AILT, con presencia de folículos linfoides reactivos debe ser interpretada con mucha prudencia y a la luz de una completa información clínica. Máxime cuando se han reportado casos de Linfoma T tipo AILT con presencia de folículos linfoides25, 26, mientras que algunos autores reportan que en estadios incipientes un linfoma AILT puede presentar folículos linfoides reactivos.4 lo cual complica aún más el espectro diagnóstico. Este caso fue evaluado por diversos anatomopatólogos de nuestra provincia y la biopsia referida para consulta a centros en Buenos Aires, USA y Japón. Sin la información clínica de buena evolución sin tratamiento, salvo por un patólogo que consideró a este proceso una hiperplasia linfoide atípica, los restantes consideraron que se trataba de un caso de Linfoma T tipo AILT, lo cual remarca la importancia de adecuada información clínica.
También la existencia entre nuestra serie, de un caso con numerosas células de carácter reactivo (granulocitos eosinófilos y células epitelioides), con una evolución clínica más prolongada que en aquellos casos con ausencia de estas células; pese a haber recibido únicamente terapia con corticoides. Parece indicar que algunos casos pueden ser precedidos de fenómenos pre-neoplásicos descriptos antes de disparar un linfoma agresivo.
Llamativo resulta que en este caso la muerte del paciente se haya producido poco tiempo después de aquella biopsia en la cual desaparece la marcada celularidad inflamatoria y se constituye finalmente un cuadro histológico típico de linfoma AILT.
Creemos importante resaltar que a nivel clínico los fenómenos observados no deben llevar a la confusión con un fenómeno reactivo. A nivel histológico la presencia de células de diámetro pequeńo con citología poco agresiva, acompańada por células inflamatorias no debe al patólogo confundirlo con un fenómeno hiperinmune. También la presencia de células de mayor diámetro con caracteres morfológicos semejantes a células de Reed-Sternberg no debe confundirse con un linfoma de Hodgkin, especialmente en aquellos casos, donde células inflamatorias acompańan a este linfoma.
Los casos con numerosas células epitelioides deben ser incluidas en el diagnóstico diferencial de ambos, linfoma de Hodgkin de la variedad a Celularidad Mixta y el linfoma de tipo Linfoepitelial o linfoma de Lennert. Otros linfomas de células T pueden presentar vénulas epitelioides prominentes, aunque no son tan marcadas como en el linfoma tipo AILT y no presentan el característico cuadro arborescente. Especialmente dificultoso puede ser el diagnóstico diferencial con linfomas T de tipo ATLL y en nuestro caso algunos de los linfomas ATLL de nuestra hospital, fueron diagnosticados como linfomas T tipo AILT, previamente a estudios para virus de HTLV-1 y fueron reclasificados luego de estos como ATLL, dando lugar a la descripción de los primeros casos de estos linfomas en nuestra provincia. En ninguno de nuestros casos se pudo documentar la transformación de éstos linfomas, en linfomas agresivos de células B como se documenta en la literatura, si bien solo en un caso se ha realizado biopsias repetidas durante la evolución de la enfermedad.
En resumen en nuestra experiencia se trata de un grupo de linfomas con un distintivo cuadro histológico (presencia de vénulas epitelioides abundantes) y expresión inmuno-histoquímica (combinación de linfocitos T CD4+ y CD8+ y nidos de células reticulares dendríticas-CD21 positivas) y con características clínicas peculiares, estadio avanzado, síntomas sistémicos y evolución agresiva.
REFERENCES
-
1) Marin O, Hasui K, Remondegui C, Sato E, Aye MM, Takenouchi N, Izumo S, Tajima K. Adult T-cell leukemia/lymphoma in Jujuy, north-west Argentina. Pathol Int 2002;52:348-357.
2) Besuschio S, Marin O, Bertoli R, Pianzola H, Diebold J, Adouin J. Découverte d´un foyer de lymphomes leucémies T de l´adulte HTLV-1 positif dans une population indienne du Nord-ouest argentin. Hématologie 2003; nş 4, vol 9 :345-347.
3) Jaffe E, Ralfkiaer E. Angioimmunoblastic T-cell Lymphoma en World Health Organization Classification of Tumours. Pathology and genetics, Tumours of the Haematopoietic and Lymphoid Tissues. Edited by Jaffe E, Harris NL, Stein H, Vardinam J. 2001; 225-226.
4) Feller AC, Diebold J. Angioimmunoblastic T-cell lymphoma. en Histopathology of Nodal and Extranodal Non-Hodgkin's Lymphomas. Based on the Who classification. Springer. 2004; 132-144.
5) Liao K, Rosai J, Daneshbod K. Malignat histiocytosis with Cutaneous involvement and eosinophilia. Am J Clin Pathol. 57:438-448.
6) Lennert K, Radaszkiewiccz T. Lymphogranulomatosis-X (LgrX). Dtsch Med Wochenschr 1975;100:1157-1163.
7) Lukes R, Tindle BH. Immunoblastic Lymphadenopathy. A hyperimmune entity resembling Hodgkin's disease. N Eng J Med. 1975;292:1-8.
8) Frizzera G, Moran EM, RappaportH. Angio-immunoblastic lymphadenopathy with dysproteinemis. Diagnosis and clinical course. Lancet. 1974;1:1070-1073.
9) Nathwani.B, Rappaport. H, Moran. E. Malignant lymphoma arising in Angioimmunoblastic lymphadenopathy. Cancer. 1978;41:578-606.
10) Shimoyama.M, Minato.K, Saito.H, y col. Immunoblastic lymphadenopathy (IBL)-like lymphoma. Jpn J Clin Oncol. 1979;9(supp):347-356.
11) Weiss L, Strickler J, Dorfman R, y col. Clonal T-cell populations in angioimmunoblastic lymphadenopathy and angioimmunoblastic-like lymphoma. Am J Pathol. 1986;122:392-397.
12) O´Connor NT, Crick JA, WaincoatJS y col. Evidence for monoclonal T-lymphocyte proliferation in angioimmunoblastic lymphadenopathy. J Clin Pathol 1986;39:1229-1232.
13) Lipford EH, Smith HR, Pittaluga S, y col. Clonality of angioimmunoblastic lymphadenopathy and implications for its evolution to malignant lymphoma. J Clin Invest. 1987;79:637-42.
14) Feller AC, Griesser H, Schilling CV, y col. Clonal gene rearrangement patterns correlate with immunophenotype and clinical parameters in patients with angioimmunoblastic lymphadenopathy. Am J Pathol 1988;133:549-556.
15) Namika R, Suchi T, Ueda R, Ito G, Koike K, Ota K, Takatsuki T. Phenotyping of proliferating in angioimmunoblastic lymphadenopathy and related lesion by the double immunoenzimatic staining technique. Am J Pathol. 1987; 127:279-287.
16) Aozasa K, Ohsawa M, Fujita M, Kanayama Y, Tominaga N, Yonezawa T, Matsubuchi T, Hirata M, Uda H, Inada E, Nakayama S. Angioimmunoblastic Lymphadenopathy: review of 44 patients with emphasis on prognostic behavior. Cancer 1989; 63:1625-1629.
17) Watanabe S, Sato Y, Shimoyama M, Minato K, Shimosato Y. Immunoblastic lymphadenopathy, angioimmunoblastic lymphadenopathy and IBL-like T-cell lymphoma: A spectrum of T-cell neoplasia. Cancer 1986;58:2224-2232.
18) E. Ralfkiaer, HK. Müller-Hermelink, E.S. Jaffe. Peripheral T-cell Lymphoma, unspecified. World Health Organization Classification of Tumours. Pathology and genetics, Tumours of the Haematopoietic and Lymphoid Tissues. Jaffe ES, Harris NL, Stein H, Vardinam J editores. pp 227-229
19) Lennert K, Feller AC. Histopathologie der Non-Hodgkin-Lymphome (nach der aktualisierten Kiel-Klassifikation). Springer-Verlag. 1990.
20) Chan JK, Banks P, Stein H, Müller-Hermelink HK, Isaacson PG, Harris NL, y col. Knowles D, Mason D, Piris MA, Ralkaier E, Pileri S, Fallini B, Jaffe E, Wranke R, De Wolff-Peeters C, Delsol G, Gatter K, Grogan T. A revised Eurpean-American Classification of the Lymphoid Neoplasms proposed by the International Lymphoma Study Group. Am J Clin Pathol. 1995:543-560.
21) Anagnostopoulos L, Hummel M, Finn T y col. Heterogeneous Epstein-Bar virus infection patterns in peripheral T-cell Lymphoma of Angioimmunoblastic lymphadenopathy type. Blood 1992;80:1804-1812.
22) Abruzzo LV, Schmidt K, Weiss L y col. B-cell lymphoma after angioimmunoblastic lymphadenopathy: a case with oligoclonal gene rearrangement associated with Epstein-Barr virus. Blood 1993;82:241-246.
23) Besuschio SC, Marin O, Bertoli R, Pianzola R, Diebold J, Audouin J. Comparación de la frecuencia relativa de las diferentes variedades de linfomas malignos no-Hodgkin en tres provincias argentinas, mostrando una prevalencia de Linfomas malignos T en ocasiones asociados a HTLV-1 en algunas poblaciones. Pren Med Argent, 2003;90:350-353.
24) Frizzera G, Moran EM, Rappaport H. Angioimmunoblastic lymphadenopathy. Diagnosis and clinical course. Am J Med. 1975;59:803-818.
25) Frizzera G. Angioimmunoblastic Lymphadenopathy en Knowles.D Neoplastic Hematopathology. Second edition. 2001. Lippincot-Willimas&Wilkins. Chapter 16. Atypical Lymphoproliferative Disorders. 2001: 569-579.
26) Ree HJ, Kadin M, Kikuchi M, y col. Angioimmunoblastic Lymphoma (AILD-Type) with hyperplastic germinal centers. Am. J Surg. Pathol. 1998; 22:643-655.
Dirección para la correspondencia: Dr. Oscar Marín.
Coronel Dávila 1027, piso 1. San Salvador de Jujuy (4600).
Argentina.
E-mail: omarin100 @ hotmail.com.
Comentario del revisor, Felix Conde MD. Hospital Can Misses. Ibiza. Espańa.
El Dr Oscar Marín nos presenta un interesante estudio en el que describe con excelente detalle las características clínico-patológicas del linfoma T angio-inmunoblástico en la provincia de Jujuy, al noroeste de Argentina. Se trata de una región geográfica con una distribución peculiar en el tipo de patología linfoide, más similar a la observada en Asia que a la observada en occidente quizá debido, sugiere el autor, a similitudes genéticas entre la población indígena de esta zona de América con la población asiática.
El estudio efectuado por el Dr. Oscar Marín y cols. contiene una interesante selección de casos de una entidad que ha generado continua controversia en Hematopatología. Algunos autores, como se menciona en el artículo, han considerado la entidad como un proceso preneoplásico o reactivo de potencial neoplásico. Esta serie apoyaría el punto de vista mayoritario de considerar la entidad como un proceso neoplásico y no reactivo, verificándose en todos estos casos un curso clínico claramente desfavorable. La caracterización de los casos se ajusta a los criterios diagnósticos aceptados, según el estado de conocimiento actual de la enfermedad. Se ha planteado que los AILT linfomas difieren fuertemente de otros linfomas de Hodgkin en sus características clínicas y de laboratorio, lo que tendría importancia pronóstica. Resulta siempre necesario enfatizar lo afirmado por los autores, acerca de la importancia para el patólogo de contar con la información clínica para correlacionarla con los hallazgos histopatológicos.
Ref: Siegert W, Nerl C, Agthe A, et al. Angioimmunoblastic lymphadenopathy (AILD) type T-cell lymphoma: prognostic impact of clinical observations and laboratory findings at presentation. Ann Oncol 1995; 6:659-664.
Es encomiable la labor de los autores en su intento por tipificar de la manera más exacta este tipo de Linfomas T, que presentan una enorme diversidad morfológica con fenotipo complejo, y es de felicitarlos por su capacidad de obtener seguimientos clínicos adecuados en sus pacientes, situación que, para los que laboramos en los trópicos, representa una tarea titánica, ya que no existe en el clínico la cultura de aportar al patólogo una historia clínica y los hallazgos del examen físico como información pertinente a cada caso.
En mi opinión, seria recomendable investigar en estos Linfomas la presencia del virus de Epstein Barr.
Agradezco la realización de este estudio tan interesante y valioso para el conocimiento de la casuística de los diferentes tipos de Linfomas en América Latina.
Recibido, 11 de marzo de 2005.
Comentario del revisor, Rodrigo Valdés Annunciata MD. Antofagasta. Chile
Comentario del revisor, Dr. Hernan Molina Kirsch Laboratorio de Patología. Ciudad de Guatemala. Guatemala.
Publicado, 11 de abril de 2005