Indice del volumen
Volume index
Comité Editorial
Editorial Board
Comité Científico
Scientific Committee
ESTUDIO DE MARCADORES BIOLOGICOS EN LA ENFERMEDAD DE ALZHEIMER
A. San Miguel, M.J. Rodríguez-Barbero, R. San Miguel, N. Alonso,
B. Calvo, FJ Martín-Gil.
Laboratorio de Análisis Clínicos. Hospital Universitario Rio Hortega.
Valladolid. Espańa
asanmiguel @ hispavista.com
Rev Electron Biomed / Electron J Biomed 2006;1:88-99
Comentario del Revisor José María Trejo Gabriel y Galán MD. PhD.. Jefe de S. de Neurología. Hospital General Yagüe. Burgos. Espańa
Comentario del Revisor Pilar Calmarza Calmarza. PhD. Bioquímica. Miguel Servet. Zaragoza. Espańa.
SUMMARY: STUDY OF BIOCHEMICAL MARKERS OF ALZHEIMER'S DISEASE.
Alzheimer´ disease (AD) affects in the World up to 25 million people and it is the most common form of dementia among older people in the Western countries. The disease usually begins after age 60, and risk goes up with age. AD can be classified as of early beginning, if it appears before the 60-65 years and of late beginning, if it appears later.
In this report, the main biological parameters in Alzheimer´disease are studied to try to differentiate it from other degenerative diseases, since its early diagnosis is of great importance.
The gene Apo E is located in the human chromosome 19 and it contains 4 exons that codes for the 299-amino acid protein named Apolipoprotein (apo) E, that is genetically polymorphic. There are three common codominant alleles, designated E2 (with a protective effect against AD), E3 (the more prevalent isoform) and E4 (which constitutes a major risk factor for AD) whose genetic basis lies within codons 112 and 158 of the gene. The three common apo E alleles lead to six common phenotypes, three homozigotes (apo E2/2, E3/3 and E4/4) and three heterozigotes (apo E3/2, E4/3 and E4/2), all originally disclosed by isoelectric focusing and immunoblotting.
As the tau it is a protein intracelular, low CSF levels can be expected. Nevertheless and by the reasons given in above paragraph, tau levels were increased in AD patients in comparison with healthy controls, especially in those having one or two Apo E4 alleles. Therefore, the test based on the quantitative determination of tau proteins in the CSF is of great help in the diagnosis of AD.
At the present time, phospho-tau is the best marker. However, it is not sensitive and specific enough to detect all the cases of AD. Therefore, the combination of Ab-42, phospho-tau and tau levels in CSF can be used as a help to confirm or exclude AD.
Key words: Alzheimer´s disease, biological markers, apolipoprotein E, apo E4, beta-amiloide, tau-protein
RESUMEN:
La enfermedad de Alzheimer afecta en el mundo a unas 25 millones de personas, siendo la causa mas frecuente de demencia en los paises occidentales. Su prevalencia va en aumento, debido al envejecimiento de la población. Por la edad de aparición la EA se puede clasificar en presenil ó de inicio temprano si aparece antes de los 60-65 ańos y senil o de inicio tardio si aparece después.
En esta revisión se estudian los principales parámetros biológicos a utilizar en la enfermedad de Alzheimer e intentar diferenciarla de otras patologías degenerativas, ya que el diagnostico precoz de la EA es de gran importancia.
El gen Apo E esta localizado en el cromosoma humano 19 y contiene 4 exones que codifica la apolipoproteina E de 299 aminoácidos. Las tres isoenzimas de la Apo E son la Apo E2, E3 y E4 y son productos de los tres alelos de cada locus génico. Tres fenotipos homozigoticos (apo E2/2, E3/3 y E4/4) y tres heterizigóticos (apo E3/2, E4/3 y E4/2) resultan de la expresión de cada uno de los tres alelos. La sustitución de los aminoácidos en los codones 112 y 158 conllevan a las diferencias entre apo E2, E3 y E4.
Como la tau se trata de una proteína intracelular el nivel hallado en LCR es bajo. El desarrollo de una elevada afinidad por los anticuerpos monoclonales altamente específicos para la tau ha conducido al desarrollo de test para la detección de la tau en LCR y un número elevado de pacientes con Alhzeimer y controles han mostrado una elevada expresión de la tau en las células neuronales afectadas. Además los test basados en la determinación cuantitativa de la proteína tau en el LCR puede ser de gran ayuda en el diagnóstico de la EA.
En la actualidad, el marcador en LCR que muestra mayor especificidad es fosfo-tau. No obstante la determinación conjunta de los tres marcadores tau, fosfo-tau y AB42 en LCR, aumenta la especificidad y sensibilidad respecto a su utilización individual.
Palabras Clave: Enfermedad de Alzheimer, demencia, marcadores biológicos, apolipoproteína E, apo E4, beta-amiloide, proteína tau.
INTRODUCCION
La enfermedad de Alzheimer (EA) es una enfermedad neurodegenerativa del SNC, asociada con una perdida de memoria progresiva que resulta en la demencia. Las dos características patológicas se observan en pacientes con EA en la autopsia: placas extracelulares y ovillos intracelulares en el hipocampo, cortex cerebral y otras áreas del cerebro esenciales para la función cognitiva.
La enfermedad de Alzheimer (EA) afecta en el mundo a unos 25 millones de personas, siendo la causa mas frecuente de demencia en los países occidentales. Su prevalencia va en aumento, debido al envejecimiento de la población, alcanzando en la actualidad cifras del 47% en personas de edad superior a los 85 ańos1.
Constituye la tercera enfermedad en casos económicos y sociales, sólo precedida por la cardiopatía isquémica y el cáncer. Es la cuarta causa de muerte en los países desarrollados tan sólo precedida por la cardiopatía, tumores e infarto cerebral2.
Atendiendo a la edad de aparición la EA se puede clasificar en presenil ó de inicio temprano si aparece antes de los 60-65 ańos y senil o de inicio tardio si aparece después. De acuerdo a si existen ó no antecedentes familiares de la enfermedad se clasifica en EA familiar ó esporádica3.
El curso natural de la enfermedad es paralelo a los procesos neuropatológicos de perdida de neuronas y sinapsis, angiopatía amiloidea, placa senil, cambio neurofibrilar de Alzheimer, etc, los cuales suceden antes de que se muestre el deterioro cognitivo de la enfermedad4. El retraso en el reconocimiento de los síntomas iniciales, unido al hecho de confusión diagnóstica con síntomas de envejecimiento, hace que el diagnostico precoz de la EA adquiera gran importancia. Para la aplicación de estrategias terapéuticas enfocadas a la prevención y al retraso de la evolución de EA, la búsqueda de marcadores biológicos potenciales para el diagnóstico precoz es fundamental.
Las placas están formadas mayoritariamente de la deposición de amiloide beta (AB) un péptido derivado de una proteína precursora amiloide (APP). Los ovillos filamentosos estan formados de filamentos helicoides apareados compuestos de neurofilamentos y proteína tau hiperfosforilada, una proteína asociada al microtúbulo. No está claro, sin embargo, si estos dos cambios patológicos son los marcadores o las causas de la EA. La EA esporádica de aparición tardía tiene una patología idéntica virtualmente a la EA familiar de aparición tempranas (FAD), lo cual sugiere por tanto un camino patológico común en ambas formas de la EA. En los estudios genéticos se han revelado 3 genes que pueden estar relacionados con formas autosómicas dominantes o EA de aparición temprana familiar (FAD). Estos genes incluyen: proteína precursora amiloide (APP), presenilina 1 (PS1), presenilina 2 (PS2) y apolipoproteína E (Apo E), pues la Apo E no origina EA por transmisión mendeliana dominante sino que aumenta la susceptibilidad y anticipa la edad de aparición. Todas las mutaciones asociadas con proteínas APP y PS pueden conducir a un aumento en la producción de péptidos AB, específicamente las formas más amiloidogenas, AB42. Además de los factores genéticos que influyen en la formación de la placa amiloide y de los ovillos intracelulares, los factores ambientales (por ejemplo citoquinas, neurotoxinas, etc) pueden también jugar importantes papeles en el desarrollo y progresión de la EA (Figura 1).
MARCADORES GENETICOS. GENES ASOCIADOS O IMPLICADOS EN EA
A pesar de que la EA se desarrolla mayoritariamente en personas mayores de 60 ańos y de forma esporádica, también existen manifestaciones precoces de la enfermedad y patrón de herencia mendeliano autosómico dominante5.
El inicio temprano de la enfermedad se relaciona con tres genes: el precursor de la proteína amiloide (APP) en el cromosoma 21, el gen presenelina-1 (Ps-1) en el cromosoma 14 y el gen presenilina 2 (PS-2) en el cromosoma 16.
No existen estudios epidemiológicos al respecto, pero mutaciones en estos genes sólo parecen explicar la mitad de las EA de inicio temprano. Es probable que existan más genes implicados. Pruebas predictivas y de diagnóstico con estos genes en EA temprana podrían ser útiles en el diagnóstico y en el consejo genético a los familiares.
El inicio tardio de la enfermedad, se asocia mayoritariamente con el gen de la apolipoproteina E (apo E) en el cromosoma 19, el cual tiene tres alelos E2, E3 y E46. La frecuencia de la variante E4 está aumentada en la EA de comienzo tardio tanto en sus forma esporádica como familiar7. La frecuencia de la variante E2 está disminuida8, 9. Como cada persona tiene dos alelos del gen pueden darse seis genotipos distintos E2/E2, E2/E3, E3/E3, E2/E4, E3/E4 y E4/E4. La presencia de apo E4 constituye un factor de riesgo para desarrollar la enfermedad y la presencia de apo E2 un factor protector. Sin embargo, existen muchas personas con EA que no tienen apo E4 y por el contrario personas con EA que con apoE4 no desarrollan la enfermedad.
La presencia del alelo E4/E4 se asocia con un comienzo más precóz de la enfermedad10, 11. La asociación entre EA y apo E4 se ha confirmado en diversos grupos étnicos12. Nos obstante, debido a que el gen apo E no es un gen determinístico sino un gen de susceptibilidad o factor de riesgo, su papel en la práctica clínica aún no está claramente determinado. Podría ser de interés la utilización como complementario a otras pruebas en el diagnostico de EA, pero no se aconseja para determinar si el indivíduo es un portador presintomático de la EA.
Estudios de ligamiento genético, apoyan la posible implicación de otros genes con EA, pero no existen confirmaciones claras. Entre ellos se puede seńalar el gen de alfa-2 macroglobulina (A2M) que se relaciona con la EA a través de la degradación del amiloide beta (AB) que se deposita en las placas seniles12, 13.
PROTEINA PRECURSORA AMILOIDE (APP)
La APP es una proteína de membrana integral, que ocurre en diferentes isoformas. Las isoformas comunes contienen 695 (APP695) 751(APP751) y 771 (APP771) aminoácidos respectivamente. Entre estas isoformas, APP695 es la principal isoforma y se expresa exclusivamente en neuronas. En contraste, APP 571 y APP770 se expresan en ambas células neuronales. La estructura primaria de APP, tiene una secuencia seńal, una region N-terminal extramembranosa grande, un dominio transmembrana simple y una cola C-terminal citoplasmática de residuo de 47 aminoácidos. Las proteínas APP maduran en el reticulo endoplasmático y en el aparato de Golgi y exhiben modificaciones post-translacionales incluyendo fosforilación, glicosilación y sulfatación.
El clivaje proteolítico de APP resulta en la generación de péptidos AB de varias longitudes. Los péptidos AB son monómeros normalmente solubles que circulan a bajos niveles en el fluido cerebroespinal y sangre. En los cerebros de pacientes con EA, la formación de placas fibrilares insolubles se facilita por un aumento y acúmulo de péptidos AB. La forma predominante de péptidos AB encontrada dentro de medio de cultivo celular condicionado y fluido cerebroespinal es el péptido AB40 más corto. AB42, sin embargo, es la forma de péptido AB inicialmente depositado dentro de las placas extracelulares del paciente con EA. Esto puede ser explicado por lo siguiente. Todas las mutaciones relacionadas identificadas dentro de APP conduce a la producción aumentada de AB42. Adicionalmente, AB42 tiende a agregarse a una velocidad más rápida y a unas concentraciones más bajas que la forma AB40.
Tres proteasas, alfa-beta y gamma secretasas estan relacionadas con el clivaje. En la superficie celular, APP sufre proteolisis por una alfa-secretasa que cliva entre Lys687 y Leu688, por tanto dando lugar a un gran ectodominio (alfa-APP). El fragmento C-terminal (83 aa, aproximadamente de 10 KDa) es retenido dentro de la membrana celular. Este fragmento puede después ser clivado por una gamma-secretasa a residuos de aminoácidos 711 o 713 dentro del dominio transmembrana dando lugar al péptido p3. Alternativamente, la superficie celular no clivada APP puede ser internalizada por las vesículas recubiertas vía endocitosis en el dominio citoplasmático distal. El APP de longitud completa puede entonces ser conducido a los lisosomas y endosomas más tardios para degradación o transferidos a endosomas prematuros para la generación de péptidos AB. En los endosomas prematuros, APP es clivado por beta-secretasa después de Met 671, creando un fragmento C-terminal retenido en una membrana (99 aminoácidos, aproximadamente de 12 KDa). El clivaje por beta-secretasa exhibe secuencia de aminoácidos primaria relativamente rígida (por ejemplo entre Met 671 y Asp 672 de APP. En la superficie de la membrana el fragmento C-terminal de 12KDa puede después ser clivado por gamma secretasa dentro del dominio de la transmembrana hidrofóbica en Va711 ó Leu713, por tanto dando lugar a un péptido AB (por ejemplo AB40 y AB42).
La identificación y caracterización de las beta y gamma-secretasas han sido áreas importantes de foco en la investigación de la EA. Aunque varios candidatos a secretasa se han sugerido la beta-secretasa BACE es la única identificada y posee actividad beta-secretasa completa. El clonaje y expresión de la enzima revela que la secretasa beta de cerebro humano BACE es una proteinasa aspártica unida a una membrana. La gamma-secretasa no ha sido definitivamente identificada trodavía. Numerosos estudios, sin embargo, han relacionado gamma-secretasa y PS1 como la misma molécula enzimática o cofactores dentro del mismo complejo.
PRESENILINA-1 (PS1) Y PRESENILINA-2 (PS-2)
La PS1 y PS2 son proteínas de membrana integrales que contienen dominios transmembrana múltiples. La PS1 y PS2 tienen una estructura predefinida similar y comparten identidad de 67% de aminoácidos. Ambas proteínas son localizadas predominantemente dentro del reticulo endoplasmático (RE) y el aparato de Golgi temprano. Son expresadas principalmente en neuronas y se expresan ubicuamente dentro del cerebro. Las proteínas PS1 y PS2 endógenas son clivadas proteoliticamente para generar 2 polipéptidos. La proteína PS1 de 46 KDa es clivada para construir un fragmento N-terminal de 28 Kda (NTF) y un fragmento C-terminal de 18 KDa (CTF), mientras la proteína PS2 de 55 KDa es clivada para construir un NTF de 35 KDa y un CTF de 20 KDa. Las especies predominantes de ambas PS1 y PS2 observadas en ambas células de mamíferos cultivadas y cerebro son los fragmentos procesados. La PS1 de longitud completa se encuentra sólo en células transfectadas y el ratón transgenico que sobreexpresa PS1.
Las funciones exactas asociadas con PS no han sido totalmente caracterizadas hoy en día. El PS1 es requerido para la formación más propia del esqueleto axial y está implicada en la neurogénesis normal y supervivencia de células progenitoras y neuronas en regiones del cerebro específicas. Las proteínas PS también se han propuesto que funcionen en el control de la apoptosis. Como se mencionó previamente, PS1 está también envuelta en la actividad gamma-secretasa. Adicionalmente, la unión de proteínas PS a APP puede jugar un importante papel en inducir la seńal intracelular.
La mayoría de los casos FAD de aparición temprana son causados por mutaciones dentro de los genes PS. Más de 40 mutaciones han sido descritos en el gen para PS1 que puede subsiguientemente resultar en FAD. Las mutaciones en ambos PS1 y PS2 son asociadas con una producción aumentada de péptido AB42 (Fig 2).
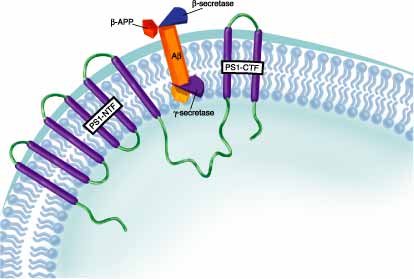
La AB42, la forma más amiloidogénica de AB, puede agregarse para formar placas amiloides difusas y neuríticas, sugiriendo que la influencia de proteínas PS en la producción de AB42 puede ser un acontecimiento inicial para el desarrollo de EA. Las mutaciones en el gen PS1 pueden también facilitar la apoptosis neuronal por desestabilización de la beta-catenina (por ejemplo parte del complejo proteína PS) predisponiendo a los individuos a la FAD de aparición temprana.
APOPROTEINA E (APO E)
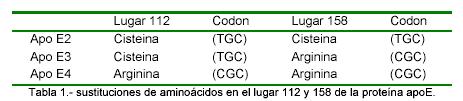
El factor de riesgo y la edad media de aparición de EA de aparición tardía está influenciada por la herencia de los alelos apo E específicos. La apo E es una proteína de 34 KDa que existe con tres isoformas principales, E2 (Cys158), E3 (Cys112 y Arg158) y E4 (Arg112). Entre las tres isoformas, Apo E3 es el más común representando aproximadamente un 78% de las formas totales, mientras que apo E4 representa el 15% y apo E2 representa el 7%. La proporción de isoformas diferentes varía entre grupos étnicos y raciales. La apo E juega un importante papel en el transporte de lípidos en sangre humana y otros fluidos corporales. Participa en el metabolismo de lipoproteínas plasmáticas, homeostasis de colesterol y procesos de transporte lipidico local. La apo E se produce por varios tipos celulares, incluyendo hígado, rińón, células grasas y macrófagos. En el cerebro, es sintetizada primariamente y secretada por astrocitos, y juega un papel principal en el transporte lipídico dentro del SNC. La presencia de la isoforma apo E4 está asociada significativamente con EA de aparición tardía. El papel exacto de la apo E4 en la patogénesis de EA no está claro. La apo E puede estar implicada en la formación de las placas amiloides u ovillos por interaccionar con proteínas tau o AB. Su expresión se considera como un factor de riesgo para la EA que no es suficiente para el desarrollo de la enfermedad.
Actualmente hay comercializado un test de la empresa Innogenetics (Inno-Lipa). El gen apo E se localiza en el cromosoma humano 19 y contiene 4 exones que codifican la apolipoproteina E de 299 aminoacidos. Las 3 isoformas principales de apo E, se refieren a como las apo E2, E3 y E4 son productos de tres alelos en un locus de gen simple. Tres fenotipos homozigoticos (apo E2/2, E 3/3 y E4/4) y 3 fenotipos heterozigóticos (apo E3/2, E 4/3 y E 4/2) surgen de la expresión de cualquiera de los 3 alelos. Los aminoácidos en los codones 112 y 158 suceden de las diferencias entre las apo E2, E3 y E4. El fenotipo más común es apo E 3/3.
La tabla 1 ilustra las sustituciones de aminoácidos en el lugar 112 y 158 de la proteina apo E.
En vista del significativo impacto diferente de los alelos de apo E en las concentraciones de colesterol LDL del plasma, la determinación del fenotipo apo E es un adyuvante importante para asegurar el perfil de riesgo cardiovascular de un individuo.
La hiperlipoproteinemia tipo III está sociada con la apo E2/2. La elevación del colesterol LDL en plasma esta relacionado con la apo E4.
La relación entre polimorfismo de apoE y EA se estableció hace ańos. La apo E4, está geneticamente asociada con las formas de enfermedad de Alzheimer familiar de aparición tardía común y esporádica.
El despistaje para establecer los factores de riesgo tales como apo E2 y apo E4 juega un papel importante como una segunda linea de diagnostico.
El Inno-Lipa apo E proporciona una herramienta para la identificación de apo E rapida y segura (Figura 3, tabla 2).

El test INNO-LiPA Apo E está basado en un principio de hibridacion inversa. El material de DNA amplificado es hibridado con oligonucleotidos especificos inmovilizados como lineas paralelas en los correspondientes tiras. Durante la amplificacion, los "primers" biotilinados son incorporados en los fragmentos amplificados de DNA. Tras la hibridación, la estreptavidina unida a la fosfatasa alcalina es ańadida y se une al hibrido biotinilado formado previamente. La incubación con un cromógeno conduce a la formación de un precipitado.
El procedimiento del test consta de los siguientes pasos:
- a) Hibridación.
En un primer paso se ańaden 10 µl de solución desnaturalizante y 10 µl de producto amplificado.
Se incuban durante 5 minutos y se coloca la tira y se incuba 30 minutos, en este caso a 45°C.
b) Lavado
Se lava las tiras y se incuban con la solución de lavado
c) Desarrollo de color
Se ańade el conjugado y se incuba y con posterioridad el sustrato y tras una nueva incubación se para la reacción con agua destilada.
MARCADORES PLASMATICOS
Existen estudios preliminares de determinaciones plasmáticas de amiloide beta y melanotransferrina (p97).
La proteína amiloide beta constituye el centro de la placa senil y se origina como resultado de la proteolisis anormal de la proteína precursora del amiloide beta14, 15. Los resultados de la determinación plasmática de amiloide B40 y B42 (AB40 y AB42) obtenidos por los distintos autores son variables, no siendo útiles en la actualidad para ayuda diagnóstica16.
Algunos autores han obtenido niveles de AB 49 y 42 superiores en pacientes con EA17 mientras que otros o bien no han encontrado diferencias significativas o han obtenido resultados contradictorios18, 19.
Existen varios estudios clínicos que relacionan la EA con alteraciones en el metabolismo del hierro y de sus proteínas. Varios autores han confirmado elevaciones séricas de la proteína p97 en pacientes afectos de EA20, 21. Se necesitan más estudios para poder avalar la utilidad de la p97 como marcador de diagnostico de EA. Sin embargo, parece que podría tratarse de un marcador incluso de fases tempranas, y además posible diferenciador de otro tipo de demencias.
MARCADORES EN LIQUIDO CEFALORRAQUIDEO (LCR)
Ya que los cambios bioquímicos en el cerebro se reflejan en el LCR, las herramientas diagnósticas en la EA se dirigen al encuentro de marcadores específicos y sensibles en dicho medio.
Los marcadores en LCR deben reflejar los procesos patogénicos centrales de dicha enfermedad, los cuales incluyen el metabolismo alterado de amiloide beta y su posterior depósito en las placas seniles y la hiperfosforilación de la proteína tau con la formación de ovillos de degeneración neurofibrilar.
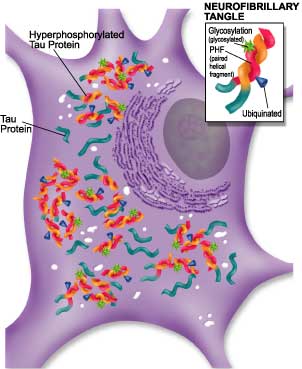
La presencia de ovillos de degeneración neurofibrilar en el citoplasma neuronal es una de las características histológicas más identificativas de la EA.. La proteína tau y la ubiquitina son las principales constituyentes de cambio neurofibrilar, estando su número directamente relacionado con la severidad de la demencia22. La agregación de tau en los ovillos se produce a consecuencia de la fosforilación irreversible, que altera la estructura de los microtúbulos e impide a la neurona trasmitir seńales eléctricas y transportar nutrientes23.
Los niveles en LCR de tau y tau fosforilada se encuentran elevados en la EA. La sensibilidad es ligeramente inferior para el marcador tau total que para tau fosforilada. Sin embargo, la especificidad es mayor para fosfo-tau24. Como se evidencia en la tabla 3, los niveles de tau y fosfo-tau en LCR están también elevados en otros desordenes neurodegenerativos y demencias.

El hecho de que la proteína amiloide B (AB) sea el principal componente de las placas seniles, hace que se estudie su utilidad como marcador biológico en LCR. La variante B42 es la que se deposita más tempranamente en las placas seniles y en varios estudios se ha mostrado una reducción de moderada a marcada en pacientes con EA frente a controles sanos25. Se han encontrado niveles bajos de AB42 en otro tipo de patologías como la demencia frontotemporal, demencia de cuerpos de Lewy y enfermedad de Creutzfeldt-Jacob (tabla 3).
En la figura 4 aparecen recogidos los marcadores biológicos en LCR utilizados en la EA y su localización.
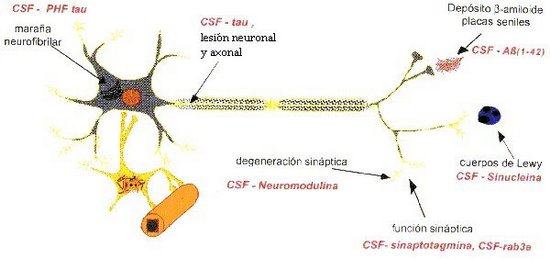
En la actualidad, el marcador en LCR que muestra mayor especificidad es fosfo-tau26, 27, 28, 29. No obstante la determinación conjunta de los tres marcadores tau, fosfo-tau y AB42 en LCR, aumenta la especificidad y sensibilidad respecto a su utilización individual.
CITOQUINAS ASOCIADAS CON EA
Las citoquinas también juegan papeles críticos en el desarrollo y progresión de EA. Las células asociadas con placas extracelulares dentro de los cerebros de pacientes con EA pueden producir una variedad de citoquinas y otras proteínas relacionadas que pueden influir intimamente en la formación de placas u ovillos. Adicionalmente, AB por si mismo puede estimular la microglia, astrositos y oligodendrocitos para secretar citoquinas proinflamatorias, quimoquinas y especies reactivas al oxígeno (ROS), las cuales pueden conducir a dańo neuronal. Las citoquinas se asocian con desarrollo y progresión de EA tales como la Il-1, Il-6, TGF-beta y TNF-alfa. Por ejemplo un perfil de expresión diferencial de varios isotipos TGF-beta puede observarse dentro de las placas de EA "ovillos neuronales" y células asociadas con placas seniles, por tanto, sugiriendo un papel para estas citoquinas que promueve el desarrollo de la lesión. La expresión de una citoquina adicional, HGF, aumenta dentro de las placas seniles, potencialmente como una función de gliosis y proliferación microglial (Fig 5)25, 26.
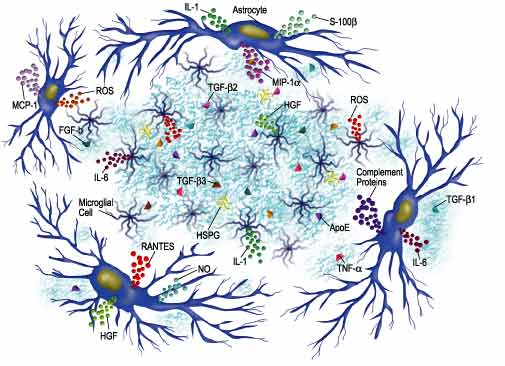
Las citoquinas asociadas tipicamente con placas amiloides (por ejemplo Il-1, Il-6 y TNF-alfa) pueden influir en la expresión de factores adicionales asociados con la patogénesis de la EA. Il-1, Il-6 y TNF-alfa pueden estimular cultivos de células neuronales y gliales in vitro, para secretar proteínas complemento. Niveles elevados de Il-1 presentes en tejido cerebral de EA pueden también influir en la expresión del factor de extensión S100beta de la neurita por astrocitos activados. La sobreregulación de S100beta puede conducir a la estimulación de crecimiento de neuritas y formación de placa neurítica finalmente. La IL-1alfa también juega un papel en la regulación de la síntesis de proteoglicano heparan sulfato (HSPG) en EA. La HSPGs son también asociadas con AB y pueden ser importantes para la agregación de péptidos AB dentro de los cerebros de pacientes con EA25, 26, 27.
CONCLUSIONES
En esta revisión hemos tratado de estudiar los parámetros biológicos principales a utilizar en la enfermedad de Alzheimer e intentar diferenciarla de otras patologías degenerativas, ya que el diagnostico precoz de la EA es de gran importancia.
El gen Apo E esta localizado en el cromosoma humano 19 y contiene 4 exones que codifica la apolipoproteina E de 299 aminoácidos. Las tres isoenzimas de la Apo E son la Apo E2, E3 y E4 y son productos de los tres alelos de cada locus génico. Tres fenotipos homozigoticos (apo E2/2, E3/3 y E4/4) y tres heterozigóticos (apo E3/2, E4/3 y E4/2) resultan de la expresión de cada uno de los tres alelos. La sustitución de los aminoácidos en los codones 112 y 158 conllevan a las diferencias entre apo E2, E3 y E4.
La presencia abundante de ambas placas seniles y marańas en el cerebro de pacientes con EA es el único criterio aceptado para un diagnóstico inequivoco de esta patología. El mayor componente de las placas seniles es un péptido llamado beta-amiloide. Este péptido se trata de un fragmento proteolítico de una proteína precursora larga conocida como proteína precursora amiloide (APP). Dado que este péptido es producido bajo condiciones normales su presencia no es especifica de EA.
Los ovillos neurofibrilares están compuestas por estructuras filamentosas helicoidales y su componente principal es una forma fosforilada anormal del microtúbulo asociado a la proteína tau. Normalmente esta proteína tiene una presencia abundante en las neuronas y sirve para estabilizar el microtúbulo en los axones. En el Alzheimer y particularmente en aquellas regiones del cerebro afectadas la proteína tau esta fosforilada de forma anormal conduciendo a un incremento en la cantidad de tau.
Como la tau se trata de una proteína intracelular el nivel hallado en LCR es bajo. El desarrollo de una elevada afinidad por los anticuerpos monoclonales altamente específicos para la tau ha conducido al desarrollo de test para la detección de la tau en LCR y un número elevado de pacientes con Alhzeimer y controles han mostrado una elevada expresión de la tau en las células neuronales afectadas. Además los test basados en la determinación cuantitativa de la proteína tau en el LCR puede ser de gran ayuda en el diagnóstico de la EA.
En la actualidad, el marcador en LCR que muestra mayor especificidad es fosfo-tau. No obstante la determinación conjunta de los tres marcadores tau, fosfo-tau y AB42 en LCR, aumenta la especificidad y sensibilidad respecto a su utilización individual.
BIBLIOGRAFIA
- 1. Flórez JA, Flórez I, Rodríguez J. Familia y enfermedad de Alzheimer: nuevos horizontes de convivencia. Med Integral 2003; 41: 178-182.
2. Llibre JJ, Guerra MA. Enfermedad de Alzheimer. Situación actual y estrategias futuras. Rev Cub Med 1999; 38: 134-142
3. López-Pousa S. Epidemiología de las demencias. En: Alberca R, López-Pousa S, eds. Enfermedad de Alzheimer y otras demencias. Madrid: IM&C 1998: 137-148.
4. Amaducci L. Impact of new therapies on Alzheimer´s disease. Management abstract book. 13th International Conference of Alzheimer´s Diseases International. Helsinki 1997.
5. Rosenberg RN: The molecular and genetic basis of AD: the end of the beginning. The 2000 Wartenberg lecture. Neurology 2000; 54:2045-2054.
6. Pérez-Tur J. La genética y la enfermedad de Alzheimer. Rev Neurol 2000; 30: 161-169.
7. Saunders AM, Strittmatter WJ, Schmechel D, St George-Hyslop PH, Pericak-Vance MA, Joo SH, et al. Association of apolipoprotein E allele E4 with late-onset familial and sporadic Alzheimer’s disease. Neurology 1993; 43: 1467-72.
8. Corder E, Saunders A, Risch N, Strittmater WJ, Schmechel DE, Gaskell PC, et al. Protective effect of apolipoprotein E type 2 allele for late onset Alzheimer disease. Nat Genet 1994; 7: 180-184.
9. Schachter F, Faure-Delanef L, Guenot F, Rouger H, Froguel P, Lesueur-Ginot L, et al. Genetic associations with human longevity at the APOE and ACE loci. Nat Genet 1994; 6: 29-32.
10. Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, et al. Gene dose of apolipoprotein E type E4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993; 261: 921-3.
11. Yoshizawa T, Yamakawa-Kobayashi K, Komatsuzaki Y, Arinami T, Oguni E, Mizusawa H, et al. Dose-dependent association of apolipoprotein E allele å-4 with late-onset, sporadic Alzheimer’s disease. Ann Neurol 1994;36:656-9.
12. Farrer LA, Cupples LA, Haines JL, Hyman B, Kukull WA, Mayeux R, y cols. Effects of age, sex, and ethnicity on the association between Apolipoprotein E genotype and Alzheimer disease. A meta-analysis. JAMA 1997; 278: 1349-1356.
13. Farińas F, Sastre I, Gil Gregorio P, Billido MJ. Enfermedad de Alzheimer: deterioro cognistivo y funcional asociado a polimorfismos de ApoE y A2M. Rev Esp Geriatr Gerontol 2002; 37: 111-119.
14. Kazze AM, Eski T, Laphan L, Gabriel K. Clinicopathologic correlates in Alzheimer´s disease: assessment of clinical and pathological diagnostic criteria. Alzheimer Dis Asocc Disord 1993; 7: 1-3.
15. Rosemberg R. A casual role for amiloid in Alzheimer´s disease: the end of the beginning. Neurology 1993;43:851-856.
16. Molinuelo JL, Lleó A, Blesa R. Marcadores diagnóstico en la enfermedad de Alzheimer. En: Martinez Lage JM, Robles Bayón A. Alzheimer 2001: Teoría y práctica. Madrid: Aula Médica eds. 2001: 93-107.
17. Tamaoka A, Fukushima T, Sawamura N, y cols. Amyloid [beta] protein in plasma from patients with sporadic Alzheimer's disease. J Neurol Sci 1996; 151:65-68.
18. Mehta P, Pirttila T, Mehta S, y cols. Plasma and cerebrospinal fluid leves of amyloid beta protein 1-40 and 1-42 in Alzheimer's disease. Arch Neurol 2000; 57:100-105.
19. Jefferies WA, Food MR, Gabathuler R, Rothenberger S, Yamada T, Yasuhara O, McGeer PL: Reactive microglia specifically associated with amyloid plaques in Alzheimer's disease brain tissue express melanotransferrin. Brain Res 1996; 712: 122-126.
20. Galasko D, Clark C, Chang L, et al. Assessment of CSF levels of tau protein in mildly demented patients with Alzheimer's disease. Neurology 1997; 48:632-635.
21. Kim DK, Seo MY, Lim SW, Kim S, Kim JW, Carroll BJ, Kwon DY, Kwon T, Kang SS. Serum melanotransferrin, p97 as a biochemical marker of Alzheimer's disease. Neuropsychopharmacology 2001; 25: 84-90.
22. Gra S, Padrón N, Llibre JJ. Péptido beta amiloide, proteína tau y enfermedad de alzheimer. Rev Cubana Invest Biomed 2002; 21: 253-261.
23. Hyslop-Greorge St PH. Piecing together Alzheimer´s. Scientific American 2000: 76-83.
24. Blennow K. CSF markers for the diagnosis of Alzheimer´s disease. Disease Focus. Alzheimer´s Disease. 2001: 1-3.
25. Agut J. Metabolismo fosfolipídico en la fisiopatología en la enfermedad de Alhzeimer. En: Acarín N, Alom J. eds. Marcadores biológicos y perpectivas terapéuticas en la enfermedad de Alzheimer. Barcelona: MCR 1989; 77-88.
26. Alom J. Neuropéptidos en la enfermedad de Alhzeimer. En: Acarín N, Alom J. eds. Marcadores biológicos y perpectivas terapéuticas en la enfermedad de Alzheimer. Barcelona: MCR 1989; 43-54.
27. Arjona A, Cano González M, Pérez Mora F, Ramos M,. Bandrés F. Enfermedad de Alzheimer. Medicina del Trabajo 1994: 3, 39-48
Esta revisión de San Miguel y colaboradores es de interés para profesionales e investigadores en neurociencias dado que responde a un problema no resuelto: el diagnóstico de la enfermedad de Alzheimer para el que no tenemos un marcador bioquímico ni de otro tipo que nos permita diagnosticarla con seguridad, rapidez y de forma temprana. Por ello los datos actuales sobre marcadores biológicos son relevantes y la investigación en este campo puede tener repercusiones muy importantes, especialmente cuando se desarrollen tratamientos de mayor eficacia que los actuales.
Esta revisión presenta interés tanto desde el punto de vista neurológico como de laboratorio. En el primer caso para establecer el diagnóstico de una enfermedad cuyo enfoque predominante es el de exclusión, es decir que es necesario descartar otras condiciones que puedan simular la enfermedad.
En el caso del laboratorio su interés radica en la ayuda que podamos prestar a los clínicos, mediante la realización de diferentes determinaciones , ya que no existe una única prueba diagnóstica que que nos permita diagnosticar la enfermedad. Muy interesante me parece,sobre todo, el párrafo referido a la asociación existente entre las citoquinas y la enfermedad de Alzheimer ya que es un aspecto poco conocido, y que pudiera ayudarnos en un futuro al diagnóstico más rápido y preciso de la enfermedad.
Recibido 21 de diciembre de 2005.
Comentario del Revisor José María Trejo Gabriel y Galán MD. PhD. Jefe de S. de Neurología. Hospital General Yagüe. Burgos. Espańa
Comentario del Revisor Pilar Calmarza Calmarza PhD. Laboratorio de Bioquímica. Miguel Servet. Zaragoza. Espańa.
* Autor para la correspondencia:
Dr. A. San Miguel Hernández
Laboratorio de Análisis Clínicos.
Hospital Universitario Rio Hortega.
Avda Cardenal Torquemada s/n
E-mail: asanmiguel @ hispavista.com
Publicado 12 de febrero de 2006.