Indice del volumen Volume index
Comité Editorial Editorial Board
Comité Científico Scientific Committee
Letters to the Editor / Cartas al Editor
CAQUEXIA DE RUSSELL ASOCIADA A ASTROCITOMA PILOCITICO: PRESENTACIÓN DE UN CASO
Leonardo Domínguez De la Ossa, Luís Rafael Moscote Salazar.
Sección de Neurocirugía. Facultad de Medicina. Universidad de Cartagena.
Cartagena de Indias, Colombia
neuromoscote @ gmail.com
Rev Electron Biomed / Electron J Biomed 2007;3:66-70
Sr. Editor:
Los diagnósticos diferenciales a considerar en nińos prepúberes con pérdida de peso y emaciación incluyen una gran cantidad de enfermedades orgánicas (enfermedades gastrointestinales, infecciones crónicas, neoplasias, desórdenes metabólicos) al igual que factores familiares y psicosociales1.
Las raras causas de emaciación profunda y falla en la ganancia de peso son los tumores que afectan el diencéfalo (especialmente la región hipotalámica y quiasmática) y aquellos que comprimen o infiltran el parénquima cerebral2-3.
Presentamos a continuación una paciente manejada por el servicio de Neurocirugía de la Universidad de Cartagena, en Cartagena de Indias, Bolívar, Colombia .
CASO CLINICO
Paciente femenino de 9 ańos de edad con cuadro clínico de 12 meses de evolución caracterizado por pérdida de peso progresiva, vómitos ocasionales y dolor abdominal frecuente e inespecífico.
Realizó múltiples ingresos a urgencias de clínica privada donde fue manejada como inapetencia.
Debido a persistencia de cuadro clínico el pediatra tratante ordenó TAC cerebral simple y contrastado que mostró lesión diencefálica e hidrocefalia, por lo que remitió a neurocirugía pediátrica.
Al examen físico la paciente se encontraba en mal estado general con signos de emaciación, peso por debajo del percentil 3 para la edad, talla conservada para la edad, con palidez mucocutánea generalizada, cardiopulmonar sin alteraciones, abdomen excavado, sin grasa subcutánea, blando, depresible, sin dolor, sin masas ni megalías, extremidades simétricas, con movilidad normal, no edemas, sistema nervioso sin déficit neurológico aparente.
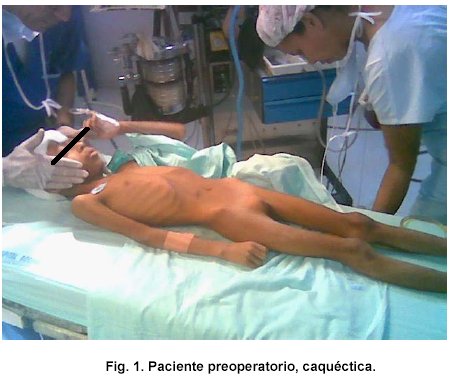
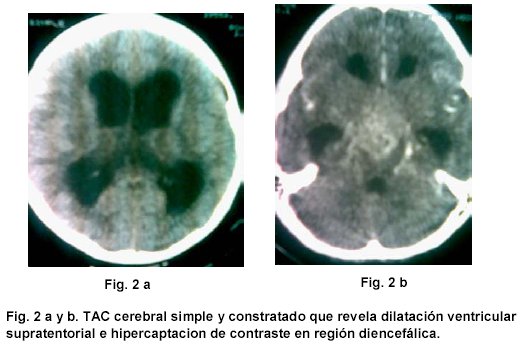
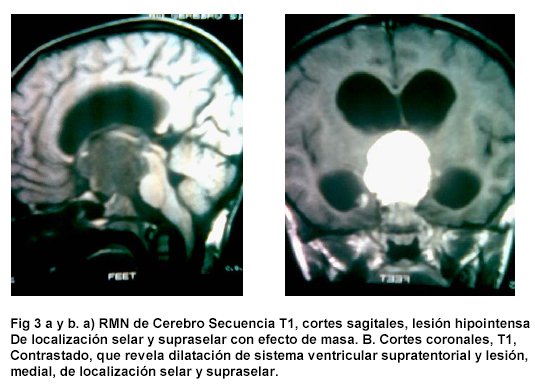
Se realizó impresión diagnóstica de síndrome diencefalico / caquexia hipotalamica de Russel y Desnutrición grado III. Se ordenó RMN cerebral con gadolinio que mostró lesión hipercaptante de localización selar y supraselar en T1.
Se decidió llevar a cirugía, realizándose abordaje subfrontal derecho y resección total de la lesión.
Al inicio de los síntomas el peso de la paciente era 25 kilogramos, al momento de la intervención quirúrgica 13 kilogramos.con posoperatorio satisfactorio, ha sido seguida en consulta externa de neurocirugía pediátrica con desaparición de signos de caquexia.
DISCUSION
En Colombia según datos del Instituto Nacional de Cancerología, los astrocitomas corresponden al 3.6 % de las neoplasias del SNC, intracranealas e intraespinales mixtas en menores de 17 ańos. El síndrome diencefálico, también conocido como Síndrome Caquectico de Russell o Enfermedad de Batten-Russell-Collier puede ser ocasionado por un Tumor diencefálico asociado con emaciación con adecuada ingesta calórica, signos y sintomas neurológicos mínimos en nińos (nistagmus, tremor, irritabilidad, hiperhidrosis)4.
Presenta una edad promedio al inicio de los síntomas de 6 a 12 meses y en menos del 4% de los casos son diagnósticados después de los 3 ańos de edad. En muchos casos existe un tumor localizado en hipotálamo, tercer ventrículo o gliomas del nervio óptico. La mayoría de tumores son gliomas y astrocitomas. Entre las características endocrinas se encuentran: pérdida de variación diurna de cortisol, aumento en los niveles basales de GH, diabetes insípida, aumento de la movilización de grasa por GH , aumento del gasto de energía, estado hipermetabólico, excesiva producción de FNTa, alteración del control del apetito. Este tipo de tumores cerebrales puede ocasionar: disfagia, vómito y alteración de la coordinación faringoesofágica; usualmente no afectan específicamente el hipotálamo lateral y puede conducir a la destrucción de hipotálamo mediobasal.
El hipotálamo es la región ventral del diencéfalo que rodea a la cavidad del tercer ventrículo. Está constituido por células neuroendocrinas. Representa el nexo entre el sistema nervioso central y el sistema endocrino. El límite anterior pasa por delante del quiasma óptico. El límite posterior pasa por detrás de los cuerpos mamilares. El límite lateral pasa por fuera de los pilares anteriores del fórnix. Ventralmente está delimitado por el tuber cinereum, que se prolonga hacia abajo en el tallo hipofisiario y el infundibulo de la neurohipófisis. La irrigación del hipotálamo esta dado asi: el principal representante arterial corresponde al círculo de Willis, pero además hay un plexo anastomótico arterial circuninfundibular y un plexo anastomótico arteriolo-capilar prequiasmático. Dentro del hipotálamo hay una rica irrigación sanguínea cerca de los núcleos neuronales. La sangre venosa hipotalámica drena en la gran vena cerebral de Galeno, vía las venas cerebral anterior, basal y cerebral interna.
El hipotálamo recibe fibras aferentes de diferentes partes del cerebro. Estas fibras pueden ser: dopaminérgicas (incerto-hipotalámica), noradrenérgicas (área tegmental), serotoninérgicas (Raphe dorsal), de galanina (tallo cerebral), glutamatoérgicas, GABA-érgicas, entre otros. El hipotálamo envía fibras eferentes a diferentes partes del cerebro así como también a la eminencia media y a la neurohipófisis.
En cuanto a la fisiología de la osmoregulacion, el hipotálamo también esta implicado. La función celular normal depende de constancia del flujo extracelular. Diariamente se pierden y se ingieren liquidos. El control de la osmolalidad plasmatica (282-298 mosm/kg) se realiza por la integración de la acción antidiurética de la vasopresina y la sensación de sed. La acción antidiurética está bajo el control del sistema endocrino hormonal, en tanto que la sensación de sed es una función del SNC. La vasopresina es liberada por la neurohipófisis en respuesta a una variedad de estímulos. Tiene un poderoso efecto antidiurético, efecto vasoconstrictor, y en los procesos de aprendizaje y memoria, más recientemente se ha descrito un rol en el estrés5-8.
Los cambios en la osmolalidad plasmática son detectadas por las neuronas del hipotálamo anterior, donde se piensa que las fenestraciones en la BHE permite el acceso de los solutos del plasma a los tejidos neurales osmosensitivos. Estudios más recientes aportan una nueva hipótesis, basada en la identificación de canales de agua (acuaporinas). En cuanto a la regulación de la temperatura podemos mencionar que el mantenimiento de la temperatura corporal depende del balance entre la producción y pérdida de calor. Este balance es regulado en el hipotálamo por 2 grupos neuronales7. La activación de los núcleos del hipotálamo anterior disminuye la temperatura corporal favoreciendo la pérdida de calor. Las neuronas localizadas en la porción posterior del hipotálamo lateral constituyen el centro de producción de calor.
Con respecto a la presión arterial el hipotálamo participa en la regulación de la misma cuando intervienen factores emocionales. La activación del hipotálamo anterior disminuye la presión arterial. La activación del hipotálamo posterior aumenta la presión arterial. Para la regulación de la ingesta de alimento el hipotálamo regula la ingesta de alimentos a través de 2 grupos neuronales conocidos como centro de la saciedad y centro del hambre. El centro de la saciedad está localizado en el núcleo ventromedial, y sus neuronas poseen receptores para la insulina. El centro del hambre está localizado en el hipotálamo lateral. Se encuentra constantemente activado, por lo que es un hecho natural el sentir hambre. El centro de la saciedad envía fibras inhibitorias al centro del hambre, de tal forma que cuando se estimula el centro de la saciedad se inhibe el hambre.
El astrocitoma pilocitico es un tumor con mejor pronóstico. Ocurren en nińos y adultos jóvenes y puede representar hasta el 28% de los tumores de fosa posterior. Úete tener la siguientes localizaciónes: cerebelo, tercer ventrículo, tálamo, hipotálamo, neurohipófisis, vía óptica anterior. Menos común: hemisferios cerebrales. Los mecanismo de las alteraciones en el crecimiento, comportamiento y metabolismo energético en la caquexia de russel son pobremente conocidos4.
Este síndrome es casi exclusivo de nińos en edades tempranas, y casi todos, tienen una lesión ocupante de espacio de la región hipotalámica - quiasmal, principalmente gliomas de bajo grado, generalmente astrocitomas pilocíticos5. Un pequeńo porcentaje (aproximadamente 6%) de los pacientes con caquexia de Russell tienen tumores de origin no glial, tales como, ependimoma, craneofaringioma y tumores de células germinales localizados en el hipotálamo anterior6. La edad promedio al inicio de los síntomas es de 6 a 12 meses. En la literatura, aproximadamente 15 % de los casos de Caquexia de Russell aparecen a la edad de 2 ańos o más6.
La falta de medro se presenta junto a una elevación en los niveles de hormona del crecimiento, lo cual, sugiere una resistencia parcial adquirida a la hormona y anormalidades en otras vías relacionadas7. La citoquina FNT a está implicada como mediador en el síndrome de caquexia por cáncer8. entre los diagnosticos diferenciales se incluyen el síndrome de Wiedemann-Rautenstrauch, sindrome de Hutchinson Gilford, la lipodistrofia parcial o síndrome glicoproteinodeficiente de carbohidratos9-10.
La importancia de este caso radica en la sospecha temprana ante un trastorno alimentario en la infancia, pues como en nuestro caso existió un sustrato tumoral que logro ser manejado de manera satisfactoria.
REFERENCIAS
- 1. Ertem, D; Acar, Y; Alper, G, et al. An Uncommon and Often Overlooked Cause of
Failure to Thrive: Diencephalic Syndrome. Case Reports. Journal of Pediatric Gastroenterology and nutrition. 2000; 30: 453-457.
2. Distelmaier, F; Janssen, G; Mayatepek,E. Disseminated pilocytic astrocytoma involving brain stem and diencephalons: a history of atypical eating disorder and diagnostic delay. J Neur Oncol. 2006; 79: 197-201.
3. Lehman RA; Krishnamurthy S, Berlin CM: Weight and height deficits in children with brain stem tumors. Clin Pediatr. 2002; 41: 315-321.
4. Murphya AM, Drummb B, Brennerc C, Lyncha SA: Diencephalic cachexia of infancy: Russell's syndrome. Case Report. Clinical Dysmorphology 2006; 15: 253-254
5. Perilongo G, Carollo C, Salviati L, et al. Diencephalic syndrome and disseminated juvenile pilocytic astrocytoma of the hypothalanmo-optic chiasm region. Cancer 1997; 80: 142-146.
6. Burr IM, Slonim AE, Danish RK, Gadoth N, Butler IJ. Diencephalic syndrome revisited. J Pediatr 1976; 88:439-44.
7. Fleischman A, Brue C, Young Poussaint Y, Kieran M, Pomeroy SL, Goumerova L, Scott RM. Diencephalic syndrome; a cause of failure to thrive and a model of partial growth hormone resistance. Pediatrics 2005; 115: 742-748.
8. Argiles JM, Busquets S, Garcia-Martinez C, Lopez-Soriano FJ. Mediators involved in the cancer anorexia-cachexia syndrome: past, present and future. Nutrition 2005;21:977-985.
9. Gropman AL, Packer RJ, Nicholson HS, et al. Treatment of diencephalic syndrome with chemotherapy, (growth, tumor response and long term control). Cancer.1998; 83:1.
10. Koelfen W, Schultze C, Varnholt V. Unusual symptoms in brain tumors inchildhood. Monatsschr Kinderheilkd. 1993;141:133-136.
Correspondencia:
Dr. Leonardo Domínguez De la Ossa.
Facultad de Medicina, Universidad de Cartagena, Campus de Zaragocilla.
E-mail: neuromoscote @ gmail.com
Recibido 14 de noviembre de 2007. Recibido revisado 27 de diciembre de 2007
Publicado 31 de diciembre de 2007