Indice del volumen Volume index
Comité Editorial Editorial Board
Comité Científico Scientific Committee
- altered cellular ATP (energy) production, which is the main one,
- altered membrane transporters
- altered cellular membrane permeability
GAUCHERīS DISEASE:
A RARE CAUSE OF FANCONI SYNDROME?
Musso CG* **, Reynaldi J*, Navarro M*, Vilas M* **,
Jáuregui R**, Imperiali N*, Algranati L*
*Nephrology Department **Centro Médico Agustín Rocca
Hospital Italiano de
Buenos Aires - Argentina
carlos.musso @ hospitalitaliano.org.ar
Rev Electron Biomed / Electron J Biomed 2007;3:46-49
Comment of the reviewer Ramón Díaz-Alersi MD. Medicina Intensiva. Hospital Puerto Real. Cádiz. Espaņa
Comment of the reviewer Abdias Hurtado Aréstegui MD. Servicio de Nefrologia. Hospital Arzobispo Loayza. Universidad Peruana Cayetano Heredia. Lima. Peru
ABSTRACT:
Gaucherīs disease consists of a genetic autosomic recesive alteration that leads to a reduction in the acid glucosil-ceramide beta-glucosidase enzyme. This enzyme brakes the glucosilceramide, a substance from which many esphingo and glucolipids are synthesized. Even though the renal compromise is not frequent in Gaucher disease, proteinuria (in nephrotic range or not) and glomerulonephritis have been described in this illness.
Fanconi syndrome is charaterized by a dysfunction in the proximal tubular reabsorption. Among the etiologies of Fanconi syndrome there are many metabolic diseases, but no association has been described yet in the literature between Fanconi syndrome and Gaucher disease. We present the following case report where this association was observed.
Key words: Gaucher disease, Fanconi syndrome, tubulopathy
RESUMEN: ENFERMEDAD DE GAUCHER. UNA CAUSA INFRECUENTE DE SINDROME DE FANCONI?
La enfermedad de Gaucher es un trastorno genético autosómico recesivo generador de un déficit de la enzima lisosomal glucosilceramida-beta-glucosidasa ácida. Dicha enzima degrada la glucosilceramida, sustancia a partir de la cual se sintetizan muchos esfingo y glucolípidos. La falta de su degradación conduce a su almacenamiento en los macrófagos tisulares con las consiguientes complicaciones mecánicas y funcionales. El compromiso renal es infrecuente en esta enfermedad, pero cuando se presenta lo hace bajo la forma de proteinuria aislada o glomerulonefritis.
El sindrome de Fanconi consiste en la disfunción parcial o total de los túbulos proximales renales. Existen diversas entidades que pueden inducir este síndrome, pero no hay hasta ahora informes en la literatura que lo vinculen con la enfermedad de Gaucher. Presentamos un caso clínico en el cual se logró documentar dicha asociación.
Palabras clave:enfermedad de Gaucher, síndrome de Fanconi, tubulopatía.
INTRODUCTION
Gaucherīs disease so called after Phillipe Gaucher who described this entity in 1882,
consists of a genetic autosomic recesive alteration that leads to a reduction in the acid glucosil-ceramide beta-glucosidase enzyme. This enzyme brakes the glucosilceramide, a substance from which many esphingo and glucolipids are synthesized1-2.
Even though the renal compromise is not frequent in Gaucher disease, proteinuria (in nephrotic range or not ) and glomerulonephritis have been described in this illness3-4.
Fanconi syndrome is charaterized by a dysfunction in the proximal tubular reabsorption which leads to an augment in the excretion of phosphate, glucose, bicorbonate, amino-acids, not necessarily all together5.
Among the etiologies of Fanconi syndrome there are many metabolic diseases such as6: Wilson disease, cistinosis, galactosemia, glucogen storage, etc; chronic metal exposition: cadmium, lead, etc. ; situations of altered immunologic state: kidney transplant, interstitial nephritis, etc; and some drugs: old tetracycline, aminoglycoside, cisplatin, etc.
Since no association has been described yet in the literature between Fanconi syndrome and Gaucher disease, we present the following case report where this association was observed.
CASE REPORT:
Male patient, 61 years old who started to be evaluated due to a picture of generalized bone pain without any other symptomathology.
As a result of his initial evaluation he was diagnosed with a spleen and hepatic enlargement. A liver biopsy showed the presence of Gaucher disease (Gaucherīs cells). An intravenous specific enzyme treatment based on imiglucerase was administered. This treatment achieved a significant reduction in the spleen and hepatic enlargement.
At the same time the above mentioned organ enlargement had been documented, a marked lumbar osteoporosis was detected in a densitometry, as well as low plasma phosphorus levels (1.8 mg/dl) in the setting of a high urinary excretion of sodium, potassium, phosphorus, calcium, magnesium, uric acid, and urea. No glucosuria and metabolic acidosis was detected (Tables I, and II).
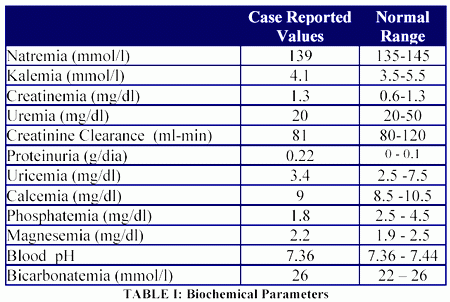
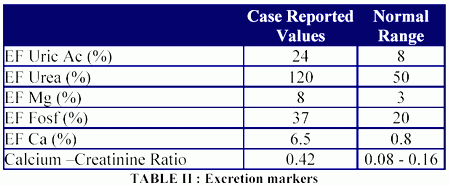
Based on the previously mentioned results, the presence of Fanconi syndrome was suspected, and due to his bone disease a monthly treatment was started using intravenous pamidronate. After this treatment he solved his bone pain and reduced, without normalizing it, his urinary calcium excretion (initial fractional excretion of calcium 6%, fractional excretion of calcium post-pamidronate infusion: 2.2%).
DISCUSSION
Gaucher disease prevails in around 1/1000 in Ashkenazi jews, while its prevalence is lower in other ethnic groups. This disease has three different clinical subtypes depending on its neurological damage2. Type I is the most frequent one (99%), it usually appears with no neuropathy and shows liver and spleen enlargement affecting either children or adults3.
All patients have their bone marrow infiltrated by macrophages filled with lipid deposits: these cells are called Gaucher's cells2.There exists bone resorption that leads to osteoporosis, spontaneous fractures and chronic pain.
At lung level there are alveolar, interstitial and pleural deposits which can lead to a cor pulmonale Kidney compromise is infrequent in Gaucher disease, but when it is present, it consist of a mild proteinuria or glomerulonephritis4.
The diagnosis is based on genetic studies in order to define the abnormal alleles. Tests and imaging also contribute to it1.
Regarding treatment it consists of symptomatic handling (analgesics) and substitutive enzyme therapy: recombined imiglucerase1.
Fanconi syndrome could cause alterations in the growth rate (children), osteomalacia (adults), water reabsorption (polyuria), hypokalemia, hypocalcemia, hypophosphatemia and tubular acidosis (type II) secondary to a reduced proximal reabsorption of sodium, potassium, bicarbonate, calcium, phosphate. Most of these patients have mild proteinuria.
Fanconi syndrome leads to urinary electrolyte loss which could be based on at least one of the three following physio-pathological mechanisms:
In this disease, the Fanconi syndrome, is produced as a consequence of the deposit of glucogen in the renal proximal tubule, interfering in the ATP generation. This syndrome is characterized by a mutation of the gen that codes the glucose 2 transporter. These patients present growing alterations, rickets, and liver and spleen enlargement5-6.
As far as we know there is no previous description in the literature of an association between Gaucher disease and Fanconi syndrome, and because of that we presented this case report where this association was observed.
Besides, since Gaucher disease is an storage disease, and it is known that some storage diseases are able to induced Fanconi syndrome through structural or functional compromise of the renal proximal tubules, perhaps Gaucher disease could occasionally induce this renal syndrome by the same mechanism.
CONCLUSION:
Gaucher disease can be associated with Fanconi syndrome, being perhaps this disease an infrequent cause of proximal renal tubular dysfunction.
REFERENCES
-
1.- Wyngaarden, Mc Govern M, Desnick R. Lysosomal storage diseases. In Bennett J, Plum F. (Eds). Cecil Textbook of Medicine. Philadelphia. Saunders Company. 1996: 1095-1099
2.- Braunwald E, Fauci A, Kasper D, Hauser S, Longo D, Jameson J. Enfermedades por depósito lisosómico. In Braunwald E, Fauci A, Kasper D, Hauser S, Longo D, Jameson J.(Eds). Harrison . Principios de Medicina Interna. MADRID. MC GRAW HILL. 2002: 2665-2666
3.- Kumar V, Abbas A, Fausto N. Genetics Disorders. In Kumar V, Abbas A, Fausto N (Eds). Robbins and Cotran. Pathologic Basis of Disease. Philadelphia. Elsevier Saunders. 2005;163-165
4.- Santoro D, Rosenbloom B, Cohen A. Gaucher disease with nephritic syndrome: Response to enzyme replacement therapy. American Journal of Kidney Disease. 2002: E4 1-4
5.- Brodehl J. Fanconi syndrome. In Cameron S, Davison A, Grünfeld JP, Kerr D, Ritz E. (Eds). Oxford Textbook of Clinical Nephrology. Oxford. Oxford University Press. 1992: 723-140
6.- Fathallah-Shaykh S, Spitzer A. Fanconi Syndrome. In Fathallah-Shaykh S, Spitzer A (Eds). Philadelphia. Medicine Wordl Medical Library. 2006: 70-75