Indice del volumen Volume index
Comité Editorial Editorial Board
Comité Científico Scientific Committee
- disfunción de transportadores de membrana,
- disturbios del metabolismo energético (reducción de la reserva de ATP),
- cambios en la permeabilidad de la membrana tubular.
ENFERMEDAD DE GAUCHER: UNA CAUSA INFRECUENTE DE SÍNDROME DE FANCONI ?
Musso CG* **, Reynaldi J*, Navarro M*, Vilas M* **,
Jáuregui R**, Imperiali N*, Algranati L*
*Servicio de Nefrología y **Centro Médico Agustín Rocca
Hospital Italiano de
Buenos Aires - Argentina
carlos.musso @ hospitalitaliano.org.ar
Rev Electron Biomed / Electron J Biomed 2007;3:50-54
Comentario del revisor Dr. Ramón Díaz-Alersi. Medicina Intensiva. Hospital Puerto Real. Cádiz. Espańa
Comentario del revisor Abdias Hurtado Aréstegui MD. Servicio de Nefrologia. Hospital Arzobispo Loayza. Universidad Peruana Cayetano Heredia. Lima. Peru
RESUMEN:
La enfermedad de Gaucher es un trastorno genético autosómico recesivo generador de un déficit de la enzima lisosomal glucosilceramida-beta-glucosidasa ácida. Dicha enzima degrada la glucosilceramida, sustancia a partir de la cual se sintetizan muchos esfingo y glucolípidos. La falta de su degradación conduce a su almacenamiento en los macrófagos tisulares con las consiguientes complicaciones mecánicas y funcionales. El compromiso renal es infrecuente en esta enfermedad, pero cuando se presenta lo hace bajo la forma de proteinuria aislada o glomerulonefritis.
El sindrome de Fanconi consiste en la disfunción parcial o total de los túbulos proximales renales. Existen diversas entidades que pueden inducir este síndrome, pero no hay hasta ahora informes en la literatura que lo vinculen con la enfermedad de Gaucher.
Presentamos un caso clínico en el cual se logró documentar dicha asociación.
Palabras clave:enfermedad de Gaucher, síndrome de Fanconi, tubulopatía.
ABSTRACT: GAUCHER´S DISEASE: A RARE CAUSE OF FANCONI SYNDROME?
Gaucher´s disease consists of a genetic autosomic recesive alteration that leads to a reduction in the acid glucosil-ceramide beta-glucosidase enzyme. This enzyme brakes the glucosilceramide, a substance from which many esphingo and glucolipids are synthesized. Even though the renal compromise is not frequent in Gaucher disease, proteinuria (in nephrotic range or not) and glomerulonephritis have been described in this illness.
Fanconi syndrome is charaterized by a dysfunction in the proximal tubular reabsorption. Among the etiologies of Fanconi syndrome there are many metabolic diseases, but no association has been described yet in the literature between Fanconi syndrome and Gaucher disease.
We present the following case report where this association was observed.
Key words: Gaucher disease, Fanconi syndrome, tubulopathy
INTRODUCCION
La enfermedad de Gaucher, llamada así en honor a Phillipe Gaucher, quien la describiese en 1882, consiste en un trastorno genético autosómico recesivo que acarrea como consecuencia el déficit de la enzima lisosomal glucosilceramida-beta-glucosidasa ácida. Dicha enzima degrada la glucosilceramida, sustancia a partir de la cual se sintetizan muchos esfingo y glucolípidos. La falta de su degradación conduce al almacenamiento de la misma en los macrófagos tisulares con las consiguientes complicaciones mecánicas y funcionales principalmente en los órganos del sistema retículo endotelial1-2.
El compromiso renal es infrecuente en la enfermedad de Gaucher, pero cuando se presenta lo hace bajo la forma de proteinuria aislada o glomerulonefritis3-4.
El sindrome de Fanconi consiste en la disfunción parcial o total de los túbulos proximales renales. Dicha disfunción puede conducir a una excesiva excreción urinaria de diversas sustancias tales como sodio, fosfato, glucosa, bicarbonato, etc. La pérdida puede involucrar a algunos de estos solutos o tan sólo a algunos de ellos5.
Entre las enfermedades potencialmente inductoras de sindrome de Fanconi se encuentran6: enfermedades metabólicas: enfermedad de Wilson, cistinosis, galactosemia, etc; exposición a metales pesados: cadmio, plomo, etc.; estados de alteración inmunológica: nefritis intersticial, transplante renal, etc. y algunos fármacos: tetraciclinas caducas, aminoglucósidos, cisplatino, etc.
Dado que no existe en la literatura una descripción de una asociación entre sindrome de Fanconi y enfermedad de Gaucher, presentamos el siguiente caso clínico en el cual dicha asociación fue documentada.
CASO CLÍNICO:
Paciente de sexo masculino, 61 ańos de edad que comenzó a ser estudiado a raíz de dolores óseos generalizados, sin otra sintomatología acompańante.
Como resultado de su evaluación se le detectó una hepato-esplenomegalia que motivó la realización de una biopsia hepática, la cual permitió el diagnóstico de enfermedad de Gaucher, por la documentación de sus típicos macrófagos cargados de lípidos (células de Gaucher). Se inició entonces tratamiento enzimático específico: imiglucerasa endovenosa, tras el cual logró una progresiva y significativa reducción de su hepato-esplenomegalia.
Concomitantemente con el hallazgo de su visceromegalia se documentó una marcada osteoporosis lumbar en una densitometría ósea, así como hipofosfatemia (1.8 mg/dl), en el contexto de una alta excreción urinaria de sodio, potasio, fósforo, calcio, magnesio, ácido úrico y urea, sin presentar sin embargo ni glucosuria ni acidosis metabólica (Tablas I y II).
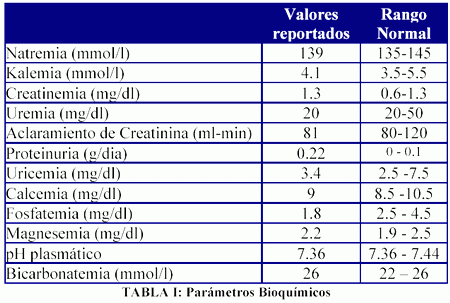
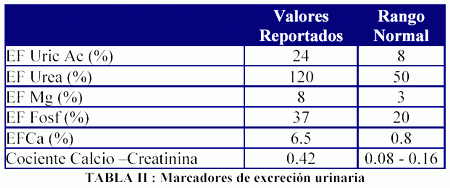
Con estos hallazgos se realizó diagnóstico de sindrome de Fanconi, y a raíz de su marcada osteopatía se inició tratamiento con pamidronato intravenoso mensual. Dicha terapéutica resolvió sus dolores óseos y redujo su excreción urinaria de calcio sin normalizarla (excreción fraccional de calcio inicial: 6% vs excreción fraccional de calcio post-pamidronato: 2.2%).
DISCUSIÓN
La enfermedad de Gaucher posee una prevalencia de 1/1000 en judíos Ashkenazi, mientras que dicha prevalencia disminuye significativamente en la población general. Esta entidad posee tres subtipos clínicos en función del dańo neurológico que presente2. El subtipo I es el más frecuente (99%), y cursa usualmente sin neuropatía y con hepato-esplenomegalia, pudiendo hallarse tanto en nińos como en adultos3.
En esta entidad es característica la presencia de infiltración de la médula ósea por macrófagos cargados de lípidos (células de Gaucher)2. Existe también marcada remodelación ósea, que puede conducir a osteoporosis, fracturas y dolores crónicos.
A nivel pulmonar, se constatan depósitos alveolares, intersticiales y pleurales, todos cambios que en suma pueden generar cor pulmonale.
El compromiso renal es infrecuente, pero cuando se presenta lo hace bajo la forma de proteinuria aislada o glomérulonefritis4.
El diagnóstico de esta enfermedad se basa en estudios géneticos (detección de los alelos anormales), aunque también contribuyen al mismo los análisis de laboratorio y estudios por imágenes1.
El tratamiento de la enfermedad de Gaucher consiste en el manejo sintomático del dolor óseo (analgésicos) y la terapia enzimática de sustitución a base de imiglucerasa recombinante1.
El sindrome de Fanconi consiste en una disfunción parcial o total de los túbulos proximales renales. Dicha disfunción puede conducir a una excesiva excreción urinaria de diversos solutos tales como sodio, potasio, aminoácidos, fosfato, calcio, glucosa, bicarbonato, etc., pudiendo entonces causar transtornos hidroelectrolíticos, acidosis tubular tipo II, retardo en el crecimiento, raquitismo (nińos) u osteomalacia (adultos).
Tres son las principales alteraciones ultra-estructurales de los túmulos proximales que pueden conducir a la instalación de un síndrome de Fanconi:
En algunas enfermedades por almacenamiento, tales como aquellas por acumulación del glucógeno, se ha reportado clásicamente asociación con sindrome de Fanconi. La entidad más característica en este sentido es el sindrome de Fanconi-Bikel, en el cual se postula que el depósito de glucógeno a nivel tubular interfiere con la generación de ATP, y por ende con el normal funcionamiento de las bómbas del túbulo proximal. Es característica de este sindrome la mutación del gen que codifica el transportador de glucosa 2. Estos pacientes presentan alteraciones del crecimiento, raquitismo, y hepato-esplenomegalia5-6.
Hasta donde sabemos no hay descripciones previas en la literatura respecto de la asociación entre enfermedad de Gaucher y sindrome de Fanconi, y por dicho motivo presentamos este caso clínico donde precisamente esta asociación ha sido documentada.
Por otra parte, dado que la enfermedad de Gaucher es una enfermedad por acumulación (tesaurismosis), y que algunas de estas entidades pueden generar sindrome de Fanconi (por ejemplo Fanconi-Bikel), a través del compromiso estructural y/o funcional tubular proximal renal, podría postularse que la enfermedad de Gaucher podría ser inductora (por supuesto infrecuente) de este sindrome renal.
CONCLUSIÓN:
La enfermedad de Gaucher puede estar asociada a sindrome de Fanconi, pudiendo tal vez esta enfermedad ser una causa infrecuente de afección tubular renal proximal.
REFERENCIAS
-
1.- Wyngaarden, Mc Govern M, Desnick R. Lysosomal storage diseases. In Bennett J, Plum F. (Eds). Cecil Textbook of Medicine. Philadelphia. Saunders Company. 1996: 1095-1099
2.- Braunwald E, Fauci A, Kasper D, Hauser S, Longo D, Jameson J. Enfermedades por depósito lisosómico. In Braunwald E, Fauci A, Kasper D, Hauser S, Longo D, Jameson J.(Eds). Harrison . Principios de Medicina Interna. MADRID. MC GRAW HILL. 2002: 2665-2666
3.- Kumar V, Abbas A, Fausto N. Genetics Disorders. In Kumar V, Abbas A, Fausto N (Eds). Robbins and Cotran. Pathologic Basis of Disease. Philadelphia. Elsevier Saunders. 2005;163-165
4.- Santoro D, Rosenbloom B, Cohen A. Gaucher disease with nephritic syndrome: Response to enzyme replacement therapy. American Journal of Kidney Disease. 2002: E4 1-4
5.- Brodehl J. Fanconi syndrome. In Cameron S, Davison A, Grünfeld JP, Kerr D, Ritz E. (Eds). Oxford Textbook of Clinical Nephrology. Oxford. Oxford University Press. 1992: 723-140
6.- Fathallah-Shaykh S, Spitzer A. Fanconi Syndrome. In Fathallah-Shaykh S, Spitzer A (Eds). Philadelphia. Medicine Wordl Medical Library. 2006: 70-75