Indice del volumen Volume index
Comitť Editorial Editorial Board
Comitť CientŪfico Scientific Committee
BŕSQUEDA DE LA MUTACI”N Phe-809 EN EL GEN DEL SUSTRATO DEL RECEPTOR DE INSULINA -1 (IRS-1) EN RATAS DIAB…TICAS eSS
Stella Maris Daniele PhD.1, Silvana Marisa Montenegro PhD.2-3,
MarŪa Cristina Tarrťs PhD.2-3, Stella Maris MartŪnez PhD 2,3.
1Facultad de Ciencias BioquŪmicas, 2Facultad de Ciencias Mťdicas, 3Consejo de Investigaciones. Universidad Nacional de Rosario.
Rosario, Argentina
smartinez948 @ yahoo.com.mx
Rev Electron Biomed / Electron J Biomed 2010;19-15.
Comentario del revisor Prof. Pilar MuŮiz RodrŪguez. Titular del Ńrea de BioquŪmica y BiologŪa Molecular de la Facultad de Ciencias de la Universidad de Burgos. EspaŮa PhD.
Comentario del revisor MarŪa Jesķs Coma MD, PhD. Head of Research Unit. Complejo Asistencial Universitario de Burgos. EspaŮa
RESUMEN
El gen del sustrato del receptor de insulina IRS-1 juega un rol clave en la transducciůn de seŮales de insulina en mķsculo esquelťtico. Diversos polimorfismos del gen IRS-1 han sido reportados en estados de insulinorresistencia como obesidad y diabetes tipo 2.
Las ratas eSS desarrollan espontŠneamente diabetes tipo 2 magra, con insulinoresistencia caracterizada por hiperglucemia e hiperinsulinemia. Durante el segundo aŮo existe disminuciůn progresiva de la insulinemia y agravamiento del sŪndrome. Nuestro propůsito fue buscar la mutaciůn Phe-809 en el gen IRS-1, que implica sustituciůn del aminoŠcido Ser por Phe en el codon 809; dicho sitio se halla conservado en la rata. Esta variante fue identificada en pacientes diabťticos tipo 2. El gen del IRS-1 consta de un solo exůn.
Se analizů un fragmento de 281pb del gen IRS-1, que posee 90% de homologŪa con el mismo fragmento en humanos. Se purificů ADN de leucocitos de un pool de sangre de ratas machos eSS y controles Wistar. Se cuantificů y amplificů por PCR utilizando un par de primers para amplificar ese mismo fragmento en el gen IRS-1 en humanos. Se usů sangre humana como control de amplificaciůn. Se empleů el mťtodo de Sanger en la secuenciaciůn del fragmento obtenido por PCR, previa extracciůn del mismo de un gel de agarosa al 2%. Se observů que la rata eSS y las controles mantuvieron la lectura esperada para ese fragmento, no encontrŠndose la mutaciůn investigada. Se descarta la mutaciůn Phe-809 como origen de la insulinoresistencia de la rata eSS.
PALABRAS CLAVE: mutaciůn Phe-809; gen IRS-1; diabetes tipo 2; ratas eSS
SUMMARY: SEARCH FOR THE PHE-809 MUTATION OF THE INSULIN RECEPTOR SUBSTRATE-1 GENE IN THE DIABETIC ESS RATS
Insulin resistance is a pathophysiological condition involved in the development of type 2 diabetes. The gene encoding Insulin Receptor Substrate - 1 (IRS-1) plays a key role in insulin-mediated signal transduction in both human and animal skeletal muscle. Several polymorphisms of IRS-1 gene have been reported to be associated with states of insulin resistance such as obesity and type 2 diabetes.
The eSS inbred-line of rats spontaneously develops a type 2 diabetes more severe in males than in females. Using eSS males, we investigated the possible presence of the Phe-809 variant in the IRS-1 gene, a site conserved in the rat, which has Ser for Phe at codon 809. This variant has been found in type 2 diabetes patients.
We analyzed a 281pb fragment of the IRS-1 gene, which has 90% homology with the same fragment of IRS-1 in humans. Leukocyte DNA was purified from a pool of blood samples of eSS rats. Wistar rats were used as controls. It was quantified and amplified by PCR with an annealing temperature of 50į C, using a pair of primers employed to amplify the same fragment in the IRS-1 gene in humans. Human blood was used as control of amplification. We employed the method of Sanger sequencing of the fragment obtained by PCR after extraction of an agarose gel 2%. When reading the radiographs, we did not find the mutation. We conclude that eSS rats do not show the Phe-809 mutation in the IRS-1 gene.
KEY WORDS: Phe-809 mutation; IRS-1 gene; type 2 diabetes, eSS rats
INTRODUCCI”N
La diabetes tipo 2 estŠ relacionada con defectos en la secreciůn de la insulina y con la resistencia a la acciůn de dicha hormona, dependiendo su expresiůn clŪnica de la interacciůn de factores genťticos y ambientales1. Tanto en pacientes como en modelos animales, la resistencia a la insulina estŠ Ūntimamente ligada al desarrollo de diabetes tipo 22-3.
En los ķltimos aŮos se han identificado en poblaciones humanas mutaciones del gen IRS-1 (sustrato de receptor de insulina) que podrŪan implicar mayor riesgo genťtico para diabetes tipo 2 al asociarse con resistencia a la insulina e hiperinsulinemia4.
Se sabe que la activaciůn del receptor de insulina (IR) induce una cascada de reacciones intracelulares que pueden estar alteradas cuando las cťlulas expresan receptores de insulina mutados, lo que se traduce en una menor acciůn de la insulina5-6. IR pertenece a una familia de molťculas receptoras de membrana con actividad tirosina quinasa. El receptor activado fosforila normalmente a una gran familia denominada sustrato del receptor de insulina (IRS), que son mediadores en la acciůn tisular de la insulina. IRS-1 fue el primer sustrato identificado y representa el prototipo del grupo IRS 7. Es el sustrato mayor para la tirosina quinasa del receptor de insulina y desempeŮa un rol clave en la transducciůn de seŮales de insulina y en el transporte de glucosa mediado por la hormona en las cťlulas de los mķsculos, el hŪgado y la grasa5,8-9.
La mutaciůn del gen IRS-1 es uno de los factores genťticos que se asocia con predisposiciůn a padecer diabetes tipo 2. En distintas poblaciones del mundo, se han detectado variaciones en el gen de IRS-1 con distinta frecuencia, algunas presentes en pacientes con insulinorresistencia que cursaban con obesidad y diabetes tipo 2 mientras que otras variantes eran silenciosas8,10.
La manipulaciůn genťtica de ratones en relaciůn con genes candidatos para resistencia a la insulina, ha permitido generar una mutaciůn nula en IRS-1 que induce un importante retardo del crecimiento, resistencia a la insulina, hiperplasia de las cťlulas beta e hiperinsulinemia aunque con sůlo una ligera intolerancia a la glucosa y sin desarrollo de diabetes manifiesta11. Posteriormente se comprobů que el alelo nulo de IRS-1 induce en los ratones una intensa resistencia a la insulina en las fibras musculares y en el hŪgado asŪ como hiperplasia marcada de la cťlulas beta mientras que ratones con la mutaciůn en el sustrato IRS-2 desarrollan intensa resistencia a la insulina en el hŪgado, moderada en el mķsculo y una modesta hiperplasia de las cťlulas beta 12.
De los polimorfismos indentificados para IRS-1 en los codones 170, 209, 809 y 97110, cuatro sustituciones de un aminoŠcido estŠn en sitios conservados tanto en humanos como en ratas y ratones9. La sustituciůn Ser809 'Phe provocarŪa alteraciones de su funciůn ya que al estar cercano a Tyr750/751 son sitios de fosforilaciůn. La sustituciůn Ser809 'Phe fue encontrada exclusivamente en pacientes diabťticos a diferencia de otras cuya prevalencia en pacientes diabťticos no discrepa de la comprobada en controles sanos10.
La rata eSS es una lŪnea con diabetes magra que manifiesta en forma temprana hiperlipidemia e incrementos moderados y progresivos hasta el aŮo de la glucemia y de la insulina plasmŠtica13-14 mientras el pŠncreas muestra imŠgenes de nesidioblastosis16. Durante el segundo aŮo de vida, sobre todo en los machos, la insulinemia disminuye con un marcado agravamiento de la hiperglucemia y una dramŠtica disminuciůn en el nķmero y tamaŮo de los islotes de Langerhans15-16.
El propůsito de este trabajo fue investigar la posible existencia de la mutaciůn Phe809 en la rata eSS.
MATERIAL Y METODOS
Se analizů en machos eSS y testigos Wistar (W) un fragmento de 281pb del gen IRS-1 que presenta un 90% de homologŪa con el mismo fragmento del IRS-1 humano.
1. Muestras: Pool de sangre de ratas machos de la lŪnea eSS y de ratas controles W de 12 meses de edad. Se utilizů sangre humana como control de amplificaciůn del fragmento (por PCR) que contiene la mutaciůn buscada.
2. Purificaciůn del ADN genůmico:
Se extrajo ADN de leucocitos de sangre obtenida por punciůn cardŪaca por la tťcnica de Proteinasa K-SDS17-19. Se determinů la concentraciůn de ADN a partir de una alŪcuota de cada uno de los ADN purificados se las diluyů en un volumen final de 1ml y se realizů un barrido de absorbancias en un espectrofotůmetro en la regiůn ultravioleta en un rango de longitudes de onda: 220-300 nm. Teniendo en cuenta que el coeficiente de absorbancia a 258nm para ADN es 50 g/ml, se calculů la concentraciůn de ADN en cada muestra. La relaciůn de absorbancias 258/280nm permitiů determinar la pureza de las muestras en cuanto al contenido de proteŪnas. La integridad del ADN se analizů por electroforesis en geles de agarosa al 1%, en presencia de buffer TBE 1x (pH 8.0), coloreados con bromuro se etidio (10mg/ml de agua) segķn tťcnicas convencionales.
3. Amplificaciůn por PCR:
Se amplificů el fragmento de 281pb, utilizando un par de primers (forward y reverse) empleados para amplificar ese fragmento del gen del IRS-1 en humanos.
Secuencia nucleotŪdica de los primers humanos usados:
- Primer forward: 5 CTCTCCTACTACTCATT
Primer reverse: 5 CAGACCAATAGCCGCCT (En la rata, en el lugar de T hay una C)
En la reacciůn de PCR se usaron 250 g de ADN genůmico, (segķn concentraciůn); 5 l de buffer 10x, 1 l MgCl2 (concentraciůn final 1,5 mM), 4 l dNTP (concentraciůn final 200 uM), 1 l primers (concentraciůn final 50 pmoles de cada uno) y 0.2 l (0.2 Unidades) GIBCO-BRL de Taq Polimerasa; se completů con H2O hasta un volumen final de 50 l. Se utilizů un termociclador Stuart Sientific teniendo como condiciones de amplificaciůn 95 ļC durante 3 min; 29 ciclos compuestos por 95ļC durante 1 min, 50ļC durante 1 min, por 72ļC durante 1min y otros 72ļC durante 5min.
El tamaŮo del producto de amplificaciůn de 281pb puede verse en Resultados.
Mediante el mťtodo de Sanger se realiza la secuenciaciůn de ADN doble cadena obtenido por amplificaciůn mediante la tťcnica de PCR. El fragmento de ADN amplificado por PCR se extrajo del gel de agarosa aL 2% despuťs de la corrida electroforťtica y para ello se usů un kit de extracciůn rŠpida del gel ("Concert Gel Extraction Systems" - Gibco BRL).
El mťtodo de secuenciaciůn desarrollado por Sanger consiste en la sŪntesis de ADN in vitro por una ADN polimerasa utilizando como templado cadena simple de ADN. La sŪntesis se inicia en un solo sitio donde un primer de oligonucleůtidos se une al templado especŪficamente, que actķa como cebador para la reacciůn de sŪntesis, ťsta se lleva a cabo por incorporaciůn de nucleůtidos complementarios, uno de ellos marcado con isůtopos radioactivos. La reacciůn termina por la incorporaciůn de un anŠlogo nucleotŪdico (dideoxinucleůtido, ddNTP) que impide la continuaciůn de la polimerizaciůn.
4. Protocolo de marcaciůn del primer:
Se realizů la marcaciůn del primer con ATP32 por medio de la enzima T4 polinucleůtido kinasa, del extremo 5 del primer especŪfico al fragmento de ADN doble cadena a secuenciar. Se utilizů como primer de secuencia (10 pmol) 4 l (diluciůn 1/60); 3 l de ATP32 (10pmol), 1 l 10x Buffer de Kinasa, 0.5 l T4 Polinucleůtido Kinasa (5U), llevando a un volumen final con agua de 10 l. Se mezclů e incubů a 37ļC por 30 min y luego se inactivů la kinasa a 90ļC por 3 min. Se centrifugů brevemente y el primer marcado se conservů a -20ļC por el tťrmino de una semana.
La mezcla de reacciůn (Mix) compuesta por 7 l de templado de ADN, 1.5 l de primer marcado con 32P, 5 l de buffer de secuencia 5x, 2.5 l de H2O esteril y 1 l de Taq Polimerasa (5u/ l). Se mezclů, se centrifugů brevemente y se conservů en hielo. Se marcaron 4 tubos de microcentrŪfuga como A, C, G y T y en cada uno de ellos se colocaron 2 l de mezcla apropiada de d/ddNTP respectivamente (mezcla de terminaciůn A, mezcla de terminaciůn C, mezcla de terminaciůn G y mezcla de terminaciůn T); se agregaron 4 l de Mix a cada tubo. Se coloců una gota de aceite mineral sobre cada mezcla y se centrifugů brevemente a 12000 r.p.m. manteniťndose el tubo en hielo.
5. Programa de ciclado para secuenciaciůn:
Se calentů el termociclador a 95ļC, se colocaron los tubos e inmediatamente se comenzů con el programa de ciclado con 95ļC por 2 minutos, luego, 30 ciclos de 30 segundos (s) a 95ļC (desnaturalizaciůn), 30 s a 58ļC (annealing) y 60 s a 70ļC (extensiůn). Una vez finalizado el ciclado se agregů a cada tubo, 3 l de soluciůn stop que contiene fosfamida y los colorantes que marcan el frente de corrida.
Previamente a la corrida electroforťtica, las muestras se desnaturalizaron a 90ļC por 5 min y se sembraron 4 l de cada reacciůn en geles de poliacrilamida al 6% con urea. Finalizada la electroforesis, el gel se fijů con Šcido acťtico al 10% y metanol al 10% por 20 min a temperatura ambiente y se seců por vacŪo. Las soluciones usadas como buffer de corrida TBE 10x, 54g Tris, 27.5 l Šcido bůrico, 20ml EDTA (0.5M) y H2O c.s.p. 500ml. La reparaciůn del gel de poliacrilamida al 6% se llevů a cabo con 126g de urea, 17.4g de acrilamida, 0.6g de bisaclilamida, 27ml de TBE 10x y H2O c.s.p. 300 ml.
Para la elaboraciůn de cada gel de secuencia se utilizaron 60ml de mezcla, 60 l de TEMED y 120 l de una soluciůn recientemente preparada de persulfato de amonio (500mg/ml). La electroforesis de 50 x 35cm de Life Technologies SA (1200 - 1500V, 60 Watts, mA libres).
Tras finalizar el recorrido electroforťtico, se realizů la radiografŪa y posterior lectura de la secuencia.
RESULTADOS
Amplificaciůn por PCR: la figura 1 muestra el recorrido electroforťtico en gel de agarosa al 2% del producto de amplificaciůn por PCR del fragmento de 281pb del gen del sustrato del receptor de insulina (IRS-1). En la calle 1 se observa el control negativo, en la calle 2 el fragmento de IRS-1 de las ratas macho eSS, en la calle 3 el fragmento IRS-1 de las ratas Wistar y en la calle 4, el fragmento de IRS-1 humano.
Secuenciaciůn del ADN doble cadena por el mťtodo de Sanger: en la posterior secuenciaciůn del producto de amplificaciůn correspondiente al fragmento de 281pb del gen del IRS-1 no se hallů la mutaciůn puntual Phe-809 (TCT' TTT) en las ratas eSS ni en las ratas controles Wistar.
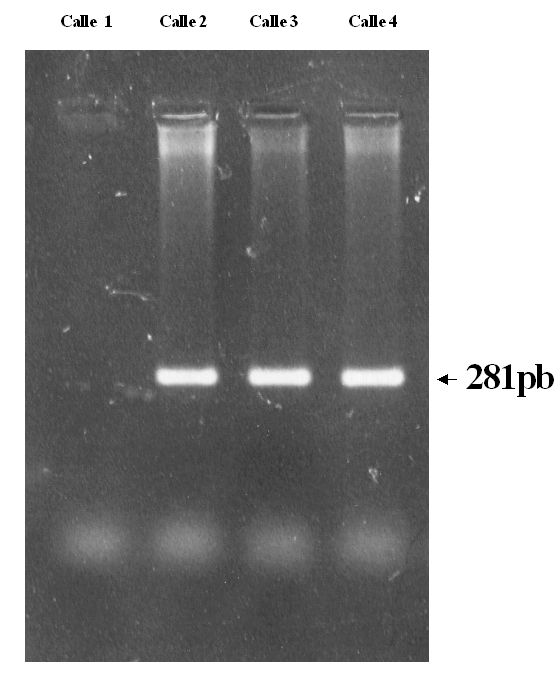
Figura 1: Recorrido electroforťtico en gel de agarosa al 2% del producto de amplificaciůn por PCR
del fragmento de 281 pb del gen del SRI -1.
Calle 1: control negativo de PCR. Calle 2: rata Wistar. Calle 3: rata eSS. Calle 4: control humano.
DISCUSI”N
Debido a la estrecha sintenia entre humanos y roedores, se ha sugerido que el estudio de diferentes cepas endocriadas puede ayudar a la identificaciůn de genes candidatos para la diabetes en seres humanos20, entre ellos los que estŠn involucrados en la seŮalizaciůn de la insulina y la insulinoresistencia21-22.
Los resultados verificados permiten concluir que la lŪnea endocriada eSS no presenta la variante Phe-809 en el gen de IRS-1, por lo que puede descartŠrsela como causante de su insulinorresistencia.
Cabe seŮalar que si bien pacientes portadores de distintas mutaciones del IRS-1 muestran anormalidades metabůlicas propias de sŪndromes de resistencia a la insulina, las mismas suelen ser marcadas8 lo que difiere del sŪndrome diabťtico de la lŪnea eSS, caracterizado por ser de moderada intensidad13. Asimismo, los modelos animales con resistencia a la insulina debida a defectos genťticos inducidos en el IRS-1 tampoco son semejantes al sŪndrome diabťtico de eSS.
Es pertinente seŮalar que la diabetes de la rata eSS surgiů en forma sķbita en la colonia de ratas eIIM de la CŠtedra de BiologŪa de la Facultad de Ciencias Mťdicas de Rosario afectando rŠpidamente a toda la descendencia13 por lo que se sugiriů como factor etiolůgico un complejo reordenamiento genťtico producto de su elevada endocrŪa, mŠs que la apariciůn sķbita de una mutaciůn puntual23. Los resultados obtenidos avalan esta hipůtesis.
AGRADECIMIENTOS:
Los autores agradecen a los doctores Gustavo Frechtel y Ariel Lůpez por sus consejos en la elaboraciůn del trabajo.
REFERENCIAS
- 1. Malecki MT. Genetics of type 2 diabetes mellitus. Diabetes Res Clin Pract 2005; 68 (Suppl1): S10-S21.
2. Reaven GM. Role of insulin resistance in human disease. Banting lecture 1988. Diabetes 1988; 37: 1595-1607.
3. Kadowaki T. Insights into insulin resistance and type 2 diabetes from knockout mouse models. J Clin Invest 2000; 106: 459-465.
4. Rung J, Cauchi S, Albrechtsen A, Shen L, Rocheleau G, Cavalcanti-ProenÁa C, Bacot F, Balkau B, Belisle A, Borch-Johnsen K, Charpentier G, Dina C, Durand E, Elliott P, Hadjadj S, Jšrvelin MR, Laitinen J, Lauritzen T, Marre M, Mazur A, Meyre D, Montpetit A, Pisinger C, Posner B, Poulsen P, Pouta A, Prentki M, Ribel-Madsen R, Ruokonen A, Sandbaek A, Serre D, Tichet J, Vaxillaire M, Wojtaszewski JF, Vaag A, Hansen T, Polychronakos C, Pedersen O, Froguel P, Sladek R R. Genetic variant near IRS1 is associated with type 2 diabetes, insulin resistance and hyperinsulinemia. Nat Genet 2009; 41: 1110-1115.
5. Sesti G, Federici M, Hribal ML, Lauro D, Sbraccia P, Lauro R. Defects of the insulin receptor substrate (IRS) system in human metabolic disorders. FASEB J 2001; 15: 2099-2111.
6. Chou CK, Dull TJ, Russel DS, Gherzi R, Lewohl D, Ullrich A, Rosen OM. Human insulin receptors mutated at the ATP-binding site lack protein tyrosine kinase activity and fall to mediate postreceptor effects of insulin. J Biol Chem 1987; 262: 1842-1847.
7. Sun XJ, Rothenberg P, Kahn CR, Backer JM, Araki E, Wilden PA, Cahill DA, Goldstein BJ, White MF. Structure of the insulin receptor substrate IRS-1 defines a unique signal transduction protein. Nature 1991; 352: 73-77.
8. Berger D, Barroso I, Soos M, Yeo G, Schafer AJ, O'Rahilly S, Whitehead JP. Genetic variants of insulin receptor substrate-1 (IRS-1) in syndromes of severe insulin resistance. Functional analysis of Ala513Pro and Gly1158Glu IRS-1. Diabetic Medicine 2002; 19: 804-809.
9. Zick Y. Role of Ser/Thr kinases in the uncoupling of insulin signaling. Int J Obes Relat Metab Disord 2003; 27 (Suppl 3): S56-60.
10. Celi FS, Negri C, Tanner K, Raben N, De Pablo F, Rovira A, Pallardo LF, Martin-Vaquero P, Stern MP, Mitchell BD, Shuldiner AR. Molecular scanning for mutations in the insulin receptor substrate-1 (IRS-1) gene in Mexican Americans with Type 2 diabetes mellitus Diabetes Metab Res Rev 2000; 16:370-377.
11. Tamemoto H, Kadowaki T, Tobe K, Yagi T, Sakura H, Hayakawa T, Terauchi Y, Ueki K, Yaburagi Y, Satoh S, Sekihara H, Yoshioka S, Horikoshi H, Furuta Y, Ikawa Y, Kasuga M, Yazaki Y, Aizawa S. Insulin resistance and growth retardation in mice made with targeted disruption of the IRS-1 gene. Nature 1994; 372: 182-186.
12. Kido Y, Burks DJ, Withers D, Bruning JC, Kahn CR, White MF, Accili D. Tissue-specific insulin resistance in mice with mutations in the insulin receptor, IRS-1, and IRS-2. J Clin Invest 2000; 105: 199-205.
13. MartŪnez SM, Tarrťs MC, Montenegro SM, Milo R, Picena JC, Figueroa N, Rabasa SL. Spontaneous diabetes in eSS rats. Acta diabetol lat 1988; 25: 303-313.
14. de Gůmez Dumm INT, Montenegro S, Tarrťs MC, MartŪnez SM, Igal RA. Early lipid alterations in spontaneously diabetic rats. Acta Physiol Pharmacol Ther Latinoam 1998; 48: 228-234.
15. Picena JC, Montenegro SM, Tarrťs MC, MartŪnez SM. Modificaciones dinŠmicas en los islotes de Langerhans de dos lŪneas de ratas espontŠneamente diabťticas: eSS y eSMT. Medicina (Buenos Aires) 2007; 67: 331-340.
16. Tarrťs MC, MartŪnez SM, Montenegro SM, Picena JC, Llorens A, Naves A. The eSS rat: A new model of non-insulin-dependent human diabetes. Am J Pathol 1992; 761-763.
17. Sambrook J, Russell DW Molecular Cloning: A Laboratory Manual. Manniatis T, Sambrook, eds. Cold Spring Harbor Laboratory Press. Third Edition. Cold Spring Harbor, NY, 2001, vol. 1, p. 1-83.
18. Davis LG, Dibner M, Battery JF. Basic methods in molecular biology, Elsevier Science Publishing, New York, 1986, p. 42-50.
19. Maniatis T, Fritsch EF, Sambrook J. Molecular cloning: A laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, 1982, p. 191-195.
20. Ktorza A, Bernard C, Parent V, Penicaud L, Froguel P, Lathrop M, Gauguier D. Are animal models of diabetes relevant to the study of the genetics of non-insulin-dependent diabetes in humans? Diabetes Metab 1997; 23 (Suppl 2): 38-46.
21. Baudry A, Loic L, Jackerott M, Joshi R. Genetic manipulation of insulin signaling, action and secretion in mice. EMBO reports 3: 323-328, 2002.
22. Nandi A, Kitamura Y, Kahn CR, Accili D. Mouse models of insulin resistance. Physiol Rev 2004; 84, 623-647.
23. Calderari S, MartŪnez SM, Tarrťs MC, Picena JC, Rabasa SL: Modelos hereditarios de diabetes y obesidad en lŪneas endocriadas vinculadas por ascendencia. Mendeliana 1995; 11: 47-55.
CORRESPONDENCIA:
Stella Maris MartŪnez.
CŠtedra de BiologŪa, Facultad de Ciencias Mťdicas
Universidad Nacional de Rosario.
Santa Fe 3100.
Rosario (2000), Argentina
E-mail: smartinez948 @ yahoo.com.mx