Indice del volumen
Volume index
Comité Editorial
Editorial Board
Comité Científico
Scientific Committee
DEVELOPMENT OF A MULTIPLE-LOCUS VARIABLE NUMBER OF TANDEM REPEAT ANALYSIS (MLVA) FOR HELICOBACTER PYLORI AND ITS APPLICATION TO HELICOBACTER PYLORI ISOLATES FROM ROSTOV REGION, RUSSIA
Vladimir M. Sorokin, Ruslan V. Pisanov, Elena A. Bereznyak, Lubov A. Prozorova.
Rostov-on-Don Institute for Plague Research
Rostov-on-Don.
Russia
Vladimir M. Sorokin soroka53 @ mail.ru
Rev Electron Biomed / Electron J Biomed 2012;3:32-39.
Comment of the reviewer Prof. Dr. José María Eirós Bouza MD, PhD. Catedrático de Microbiología de la Facultad de Medicina de la Universidad de Valladolid. Valladolid. Espańa.
Comment of the reviewer Dra. María Ángeles Mantecón, PhD. Department of Microbiology. Hospital Universitario de Burgos. Burgos. Espańa
SUMMARY:
Stomach infection with Helicobacter pylori (H. pylori) is the second most common infectious disease of humans.
The severe pathological consequences of this infection include gastric and duodenal ulcer disease, the development of gastric mucosal atrophy, gastric carcinoma, and, more rarely, malignant tumors of the lymphoma. H. pylori infections cause very high morbidity and mortality and are of particular concern in developing countries, where H. pylori prevalences as high as 90% have been reported.
The population of H. pylori shows a high genomic variability among isolates. And the polymorphism of repeat-units of genomics had participated the important process of evolution. A variety of molecular typing tools have been developed to access genetic relatedness in H. pylori isolates. However, there is still no standard genotyping system of this bacterium.
The MLVA (Multi-Locus of Variable number of tandem repeat Analysis) method is useful for performing phylogenetic analysis and is widely used in bacteria genotyping; however, there's little application in H. pylori analysis.
This article is the first application of the MLVA method to investigate H. pylori isolates in Russia. MLVA of 4 VNTR loci with high discrimination power based on 10 candidates were performed on a collection of 22 strains of H. pylori which originated from Rostov region of Russia. This method provides a starting point on which improvements to the method and comparisons to other techniques can be made.
KEYWORDS: Helicobacter pylori. VNTR genotyping. Cluster analysis
RESUMEN: DESARROLLO DE UN "MULTIPLE-LOCUS VARIABLE NUMBER OF TANDEM REPEAT ANALYSIS" (MLVA) PARA HELICOBACTER PYLORI Y SU APLICACIÓN AL HELICOBACTER PYLORI AISLADO EN LA REGIÓN DE ROSTOV, EN RUSSIA.
La infección del estómago por Helicobacter pylori (H. pylori) es la segundo más común de las enfermedades infecciosas humanas.
Las graves consecuencias patológicas de esta infección incluyen úlcera gástrica y úlcera duodenal, el desarrollo de atrofia de la mucosa gástrica, cáncer gástrico, y, más raramente, tumores malignos de tipo linfoma. Debido a la muy elevada morbilidad y mortalidad del H. pylori es de especial preocupación en los países en desarrollo, donde se han reportado prevalencias de H. pylori de hasta el 90%.
La población de H. pylori muestra una alta variabilidad genómica entre las diversas cepas. El polimorfismo de repetición en las unidades genómicas ha participado en el importante proceso de evolución. Una variedad de herramientas de caracterización molecular se han desarrollado para acceder a la caracterización genética de cepas aisladas de H.pylori. Sin embargo, existe todavía ningún sistema estándar de determinación del genotipo de esta bacteria.
El método MLVA (Multi-Locus of Variable number of tandem repeat Analysis) es útil para llevar a cabo análisis filogenéticos y se utiliza ampliamente en el procedo de genotipado de bacterias, sin embargo, es escasa su aplicación en análisis de H. pylori.
Este artículo describe la primera aplicación del método MLVA para investigar muestras de H. pylori aisladas en Rusia. MLVA de cuatro VNTR loci, con alto poder de discriminación basado en 10 candidatos se realizó en una colección de 22 cepas de H. pylori procedentes de la región de Rostov de Rusia. Este método proporciona un punto de partida susceptible de mejora y comparacion con otras tecnicas.
PALABRAS CLAVE: Helicobacter pylori. Genotipado VNTR. Analisis de cluster.
INTRODUCTION
H. pylori isolates obtained from different individuals and ethnic groups in the world exhibit substantial genomic diversity due to synonymous substitutions, insertion-deletion (indel)polymorphisms, and mobility of repetitive elements. This diversity could be further enhanced by chromosomal rearrangements due to a high level of interstrain recombination. Geographical partitioning of the gene pool exists within H. pylori, and sequences are less related between isolates from different populations than between isolates from families1.
Several molecular typing tools were tried for strain typing and identification of H. pylori isolates. These include pulsed-field gel electrophoresis2, random fragment length polymorphism3, randomly amplified polymorphic DNA4, 5, amplified fragment length polymorphism6,7, and PCR-based genotyping of repetitive sequences, namely, repetitive extragenic palindromes8,9 and enterobacterial repetitive intergenic consensus elements10.
All these techniques indicate that the H. pylori population genetic structure is panmictic, and a high level of DNA diversity is found within strains. However, all of these methods suffer from one or more significant drawbacks, including insuficient discriminatory power, poor reproducibility between laboratories, and dificulties with the comparison and accumulation of results by different laboratories.
As an alternative to the above methods, investigation of Variable Number of Tandem Repeats (VNTR) has been described for various organisms. These include Salmonella enterica11, Staphylococcus aureus12, Yersinia pestis13, Mycobacterium tuberculosis14, Francisella tularensis15 and others.
VNTR are repeated DNA sequences of varying copy number. They are caused by slipped strand mispairing during DNA replication16,17. VNTRs can provide information relating to both the evolutionary and functional areas of bacterial diversity16. The ability to detect VNTRs in microorganisms has been greatly enhanced by the availability of whole genomic sequences and software that can search for VNTR loci from these sequences18. Once these polymorphisms are located, flanking primers can then be designed to amplify these variable length regions thus allowing differentiation of copy numbers using the size of the resultant amplicon. This can be done using standard agarose gel electrophoresis and if a higher resolution is required, polyacrylamide gel electrophoresis.
VNTR is therefore applicable to a wide range of laboratories, including those which may have simple equipment such as thermal cyclers and agarose/ polyacrylamide gel electrophoresis but do not have access to sophisticated equipment such as DNA sequencers.
Furthermore when VNTR is applied to multiple loci as a typing scheme such as in Multiple Locus VNTR Analysis (MLVA) greater discriminatory power and more accurate determination of genetic relatedness is achieved13,15,19,20.
In this paper, we report on the development of a MLVA scheme using novel VNTR loci selected from the sequences of a published H. pylori genome and evaluate its usefulness as a typing method using reference strain and clinical isolates from Rostov region of Russia.
MATERIAL AND METHODS
Bacterial strains and DNA isolation.
Reference strain H.pylori NCTC 11637(Russia) was used in this study. Twenty-two H. pylori isolates were recovered from antral gastric biopsies of patients. For culture, biopsy samples were homogenized and inoculated onto Trypticase soy agar plates supplemented with 7.5% sheep blood. Cultures were identified by urease, catalase, and oxidase tests and Gram staining. Genomic DNA was extracted from bacterial isolates using a QIAamp DNAMini Kit (Qiagen), according to the manufacturer's instructions. DNA was eluted in 200 µL of elution buffer and 5 µL of each DNA solution was used in the PCR.
VNTR primer design.
DNA sequences of three H. pylori strains (26695, J99 and G27) deposited in GenBank under accession numbers, NC000915, NC000921 and NC011333 were used to detect the VNTR loci. Analysis using the Tandem Repeat Finder (TRF) program http://tandem.bu.edu/18 was used to identify potential VNTR loci. Primer Premier 5.0 (Premier Biosoft) was used to design PCR primers for amplifying the loci. Primers were designed within the flanking regions, with a theoretical melting temperature of 57°C to 60°C.
VNTR PCR amplification.
A PCR reaction mixture (30 µL) containing 5 µL of DNA template, 10 pmol of each primer, 1 unit of Taq DNA polymerase, 200 µM of dNTPs and 10 x PCR buffer (500 mM KCl, 100 mM TrisHCl (pH 8.3) 25 mM MgCl2) was utilized. Amplification was carried out in a DNA thermocycler Tercyc (DNA-Technology, Russia) with denaturation at 94°C for 5min, followed by 30 cycles of denaturation at 94°C for 45 s, annealing at 58°C for 45 s and elongation at 72°C for 1 min.
A 5-min elongation at 72°C was performed after the last cycle to ensure complete extension of the amplicons. Each PCR product (5 µL) was resolved by 5-8% polyacrylamide gel electrophoresis with 1X TBE (90 mM Tris-borate, 1 mM EDTA, pH 8). Allelic sizes were estimated using a pBlueScript DNA / MspI (MBI Fermentas, Vilnius, Lithuania) as a size marker. Gels were visualised using UV transillumination and the images captured using the ChemiDoc XRS System (BioRad).
DNA sequence analysis.
The PCR products from reference strain were sequenced using the same primers used to amplify the products. Sequencing reactions were performed using the BigDye terminator technology according to the manufacturer's recommendation (Applied Biosystems), and products were analyzed in an ABI 3130 capillary electrophoresis system equipped with the POP 4 matrix (Applied Biosystems). Data obtained with forward and reverse sequencing primers were combined, and sequences were manually aligned.
Data Analysis.
Using the Quantity One 1D Analysis software package (BioRad), the polyacrylamide gel images were analysed and allelic sizes estimated. Allelic sizes were then converted into repeat copy numbers using the formula: Number of Repeats (bp) = [Fragment size (bp) - Flanking regions (bp)] / Repeat size (bp). The repeat copy numbers were then rounded down to form whole numbers. When repeat numbers were less than one, they were rounded down to zero, whilst no amplification was represented by the number ninety-nine. This created a numerical profile which was analysed as a character dataset using Statistica software package version 6.0. Clustering analysis was done using the categorical parameter and the Ward coefficient. Nei's Diversity Index of the VNTR loci was calculated from the range of alleles generated from the strains used utilising the formula;
D = 1-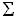 (allele frequency)2 (Weir, 199021).
(allele frequency)2 (Weir, 199021).
RESULTS AND DISCUSSION
Earlier, we have described a MLVA typing scheme based on 3 VNTR loci for differentiation of H. pylori clinical isolates22. The disadvantage of previous scheme was inability to detect any amplicons for several H. pylori strains.
The repeat sequence of loci from 7 to 12 bp and consistency of repeat unit > 90% were selected for this research to apply polyacrylamide gel electrophoresis for more accurate determination of bands size. In this study, 10 VNTR loci were candidated from the H. pylori database.
And we finally identified 4 VNTR highly polymorphic loci and designed primers for their detection (Table 1).
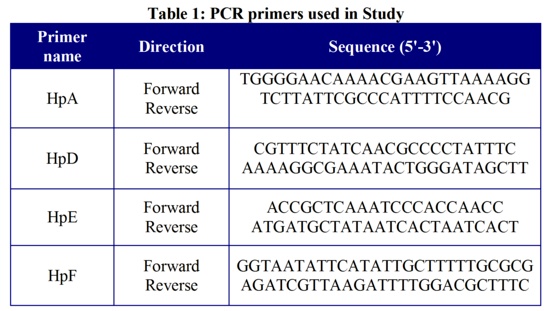
The remaining 6 loci either failed to amplify any DNA or were amplified but were monomorphic. The main characteristics of the 4 VNTR loci are listed in Table 2, including the diversity index of each locus.
Clustering analysis revealed two major clusters and allowed to obtaine 24 individual genotypes, including reference strain and predicted data for H. pylori J99 (data not shown). In addition, we tested possibility to detect VNTR,s directly in biopsies and observed positive results in 10 from 15 samples studied.
This preliminary investigation validates a first set of markers for MLVA investigation of H. pylori clinical isolates. The loci used in this study provided high discriminatory power and suc- cessfully separated closely related isolates of different strains from Rostov region of Russia.
The limitation of the most MLVA assays described is the use of agarose gel electrophoresis to separate fragments for allelic sizing, due to inherent inaccuracies of this method to size bands of close molecular weights. So, we used polyacrylamide gel electrophoresis to receive a higher resolution and accurate determination of repeats number.
In contrast with other genotyping methods, the relatively low cost and moderate expertise required for MLVA typing would allow the systematic typing of any new isolate directly by clinical laboratories. All markers proposed here are easy to type with no sophisticated equipment and software.
The usefulness of the MLVA typing scheme proposed here must be further determined by investigating a larger population of isolates from Russia and other countries.
Further improvements need to be made to the method so that MLVA can be applied directly to biological (biopsies, faeces) and environmental samples, thus avoiding culturing of the pathogen. This would allow epidemiological studies in developing countries where it is not always possible to culture H. pylori .
REFERENCES
1.- Achtman M, Azuma T, Berg DE, Ito Y, Morelli G, Pan ZJ, Suerbaum S, Thompson SA, van der Ende A, van Doorn LJ. Recombination and clonal groupings within Helicobacter pylori from different geographical regions. Mol Microbiol. 1999; 32:459-470.
2.- Takami S, Hayashi T, Akashi H, Shimoyama T, Tamura T. Genetic heterogeneity of Helicobacter pylori by pulse-field gel electrophoresis and re-evaluation of DNA homology. Eur J Gastroenterol Hepatol. 1994; Suppl.1:S53-S60.
3.- Salaun L, Audibert C, Le Ley G, Burucoa C, Fauchere JL, Picard B. Panmictic structure of Helicobacter pylori demonstrated by the comparative study of six genetic markers. FEMS Microbiol Lett.1998;161:231-239.
4.- Owen RJ, Bickley J, Costas M, Morgan DR. Genomic variation in Helicobacter pylori: application for identification of strains. J Gastroenterol. 1991;181:43-50.
5.- Akopyantz N, Bukanov NO, Westblom TU, Kresovich S, Berg DE. DNA diversity among clinical isolates of Helicobacter pylori detected by PCR-based RAPD fingerprinting. Nucleic Acids Res. 1992; 20:5137-5142.
6.- Gibson JR, Slater E, Xerry J, Tompkins DS, Owen RJ. Use of an amplified-fragment length polymorphism technique to fingerprint and differentiate isolates of Helicobacter pylori. J Clin Microbiol. 1998;36:2580-2585.
7.- Ahmed N, Khan AA, Alvi A, Tiwari S, Jyothirmayee CS, Kauser F, Ali M, Habibullah CM. Genomic analysis of Helicobacter pylori from Andhra Pradesh, South India: molecular evidence for three major genetic clusters. Curr Sci. 2003;85:1579-1586.
8.- Miehlke S, Thomas R, Guiterrez O, Graham DY, Go MF. DNA fingerprinting of single colonies of Helicobacter pylori from gastric cancer patients suggests infection with a single predominant strain. J. Clin.Microbiol. 1999;37:245-247.
9.- van Doorn NE, Namavar F, van Doorn LJ, Durrani Z, Kuipers EJ, Vandenbroucke-Grauls CM. Analysis of vacA, cagA, and IS605 genotypes and those determined by PCR amplification of DNA between repetitive sequences of Helicobacter pylori strains isolated from patients with nonulcer dyspepsia or mucosa-associated lymphoid tissue lymphoma. J Clin Microbiol. 1999;37:2348-2349.
10.- Thoreson AC, Hosseini N, Svennerholm AM, Bölin I. Different Helicobacter pylori strains colonize the antral and duodenal mucosa of duodenal ulcer patients. Helicobacter 2000;5:69-78.
11.- Ramisse V, Houssu P, Hernandez E, Denoeud F, Hilaire V, Lisanti O, Ramisse F, Cavallo JD, Vergnaud G. Variable Number of Tandem Repeats in Salmonella enterica subsp. enterica for Typing Purposes. J Clin Microbiol 2004, 42:5722-5730.
12.-Sabat A, Krzyszton-Russjan J, Strzalka W, Filipek R, Kosowska K, Hryniewicz W, Travis J, Potempa J. New method for typing Staphylococcus aureus strains: multiple-locus variable-number tandem repeat analysis of polymorphism and genetic relationships of clinical isolates. J Clin Microbiol 2003, 41:1801-1804.
13.- Adair DM, Worsham PL, Hill KK, Klevytska AM, Jackson PJ, Friedlander AM, Keim P: Diversity in a variable-number tandem repeat from Yersinia pestis. J Clin Microbiol 2000, 38:1516-1519.
14.- Frothingham R, Meeker-O'Connell WA. Genetic diversity in the Mycobacterium tuberculosis complex based on variable numbers of tandem DNA repeats. Microbiology 1998, 144 (Pt 5):1189-1196.
15.- Farlow J, Smith KL, Wong J, Abrams M, Lytle M, Keim P: Francisella tularensis strain typing using multiple-locus, variable-number tandem repeat analysis. J Clin Microbiol 2001;39:3186-3192.
16.- van Belkum A, Scherer S, van Alphen L, Verbrugh H. Short-sequence DNA repeats in prokaryotic genomes. Microbiol Mol Biol Rev 1998, 62:275-293.
17.- Bzymek M, Lovett ST. Instability of repetitive DNA sequences: the role of replication in multiple mechanisms. Proc Natl Acad Sci USA. 2001, 98:8319-8325.
18.- Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res 1999; 27:573-580.
19.- Keim P, Price LB, Klevytska AM, Smith KL, Schupp JM, Okinaka R, Jackson PJ, Hugh-Jones ME. Multiple-locus variable-number tandem repeat analysis reveals genetic relationships within Bacillus anthracis. J Bacteriol 2000; 182:2928-2936.
20.- Klevytska AM, Price LB, Schupp JM, Worsham PL, Wong J, Keim P. Identification and characterization of variable-number tandem repeats in the Yersinia pestis genome. J Clin Microbiol 2001, 39:3179-3185.
21.- Weir BS. Genetic data analysis: methods for discrete population genetic data analysis. Sinauer Associates Inc., Sunderland, Mass. 1990.
22.- Sorokin V, Pisanov R, Vodopyanov A. VNTR-genotyping analysis of Helicobacter pylori clinical isolates. Zdravoohranenie RF 2011; 5:45-46.
CORRESPONDENCE:
Vladimir M. Sorokin
Rostov-on-Don Institute for Plague Research,
344007, M. Gorkogo 117,
Rostov-on-Don
Russia
Mail:soroka53 @ mail.ru