Indice del volumen Volume index
Comité Editorial Editorial Board
Comité Científico Scientific Committee
SÍNDROME DE ALPORT.
I.- FISIOPATOLOGÍA Y CLÍNICA
Natalia Zabala MD.
Sanatorio de la Trinidad Palermo.
Buenos Aires, Argentina
natalialzabala @ hotmail.com
Rev Electron Biomed / Electron J Biomed 2016;1:51-57.
Comentario del revisor Carlos G. Musso, MD. PhD. Profesor Asociado de Fisiología Humana. Instituto Universitario del HIBA. Jefe de Fisiología Clínica Renal y de la Unidad de Diálisis Peritoneal. Hospital Italiano de Buenos Aires (HIBA). Argentina
Comentario del revisor Prof. Juan F. Macías-Núńez, MD. PhD.
Servicio de Nefrología. Hospital Universitario de Salamanca. Espańa
RESUMEN
El síndrome de Alport es una enfermedad hereditaria, usualmente ligada al cromosoma X, que se caracteriza por presentar nefropatía progresiva, sordera y oftalmopatía, y se basa en una alteración en el colágeno tipo IV.
PALABRAS CLAVE: Alport. Nefropatia.
SUMMARY:
Alport syndrome is an inherited disease, usually linked to the X chromosome, which is characterized by progressive nephropathy, deafness and eye disease, and is based on an alteration in type IV collagen.
KEY WORDS: Alport. Nephropaty.
INTRODUCCIÓN
Cecil A. Alport, fue un médico británico, que en 1927 describió 3 generaciones de una familia con combinaciones de nefritis hereditaria progresiva y sordera. El Dr. Alport también observó que la microhematuria fue el síntoma más común y que los hombres se vieron afectados más severamente que las mujeres1.
Su reconocimiento de esta asociación y la descripción de más familias con dichos trastornos, llevaron a que esta enfermedad fuera nombrada Síndrome de Alport (SA) en el ańo 19611,2.
En la actualidad se la describe como una enfermedad hereditaria que afecta a las membranas basales, causada por alteraciones en una de sus proteínas estructurales: el colágeno tipo IV1-3.
Se estima que su prevalencia en la población general es de 1 cada 50.000 habitantes4.
Patrones de Herencia
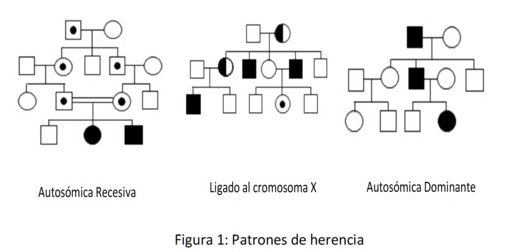
Se transmite mediante tres patrones de herencia diferentes: la herencia ligada al cromosoma X, la herencia autosómica recesiva y la herencia autosómica dominante. Comparten características clínicas comunes pero la historia familiar varía para cada uno de ellos1-3.
Síndrome de Alport ligado al cromosoma X: SALX:
El SA se caracteriza por: hematuria, proteinuria significativa, hipertensión, sordera neurosensorial y progresión hacia insuficiencia renal crónica terminal (IRCT). Estos síntomas son generales a todas las formas de SA, pero en el caso del SALX se limitan únicamente a aquellos pacientes que sean hombres, como toda enfermedad ligada al cromosoma X sólo los hombres la padecen y las mujeres son portadoras. Sin embargo, las mujeres portadoras de SALX tienen una clínica muy variable (desde enfermedad asintomática hasta una presentación severa)
La variabilidad en la expresión de la enfermedad en mujeres puede explicarse por el fenómeno de inactivación del cromosoma X o lionización, que tiene lugar en las mujeres para alcanzar una compensación de dosis. Durante el desarrollo embrionario, uno de los dos cromosomas X de cada célula es inactivado permanentemente y al azar, sobre todo mediante procesos de metilación del ADN. El patrón de inactivación en cada célula progenitora se transmite con una alta estabilidad a sus células descendientes. Por tanto, es de esperar una tasa de 1:1 entre las células que expresen el cromosoma X normal y las que expresen el cromosoma X que lleva la copia del gen COL4A5 mutada.
Las mujeres portadoras de SALX con una clínica grave serán las que hayan sufrido una inactivación preferencial del cromosoma X normal en sus células embrionarias y, por tanto, expresen en mayor proporción el cromosoma X con el gen COL4A5 mutado. De la misma manera, habrá mujeres portadoras que sean completamente asintomáticas por haber tenido una inactivación preferencial del cromosoma X que lleva la mutación. Mutaciones en el gen COL4A5 son la base molecular del SALX que representa el 80-85% de los casos familiares de SA. Existe una correlación significativa entre la edad de inicio de la IRCT y el genotipo de los pacientes. Las mutaciones más importantes suelen dar lugar a formas más graves de SALX. Esto es mucho más evidente en hombres que en mujeres (Figura 1).
Síndrome de Alport autosómico recesivo: SAAR:
El SAAR se caracteriza por los síntomas clásicos de SA, pero éstos están presentes por igual en hombres y en mujeres. La prevalencia y el patrón clínico en los individuos portadores se están aún por determinar, aunque la hematuria parece ser uno de los síntomas mayoritarios. El SAAR debe sospecharse cuando un individuo exhibe el cuadro clínico y patológico de la enfermedad pero carece de antecedentes familiares, especialmente cuando una mujer posee síntomas indicativos de enfermedad grave como sordera, insuficiencia renal o proteinuria severa en la juventud. La sospecha de un patrón de herencia autosómico recesivo debe ser especialmente fuerte cuando se dé consanguinidad entre los padres (Figura 1).
Síndrome de Alport dominante: SAAD:
Clínicamente es igual al SAAR (es decir, un SA que se puede presentar tanto en varones como en mujeres) pero con una herencia autosómica dominante. Además, se postula que en los casos de SAAD la progresión a IRCT pudiera ser más lenta que en otros tipos de SA. Para aceptar este patrón de herencia se exige una transmisión varón-varón de la enfermedad (Figura 1).
Genética y desarrollo del colágeno tipo IV
El colágeno tipo IV fue aislado por primera vez de la MBG canina en 1966.
La familia de proteínas del colágeno tipo IV está formada por seis isotipos, designados α1 (IV) - α6 (IV). Las seis cadenas tienen alta homología en la secuencia. Tienen tres dominios: un dominio 7S corto en el N-terminal de 15 a 20 residuos; un dominio largo de colágena que ocupa la sección media de la molécula, de alrededor de 1 400 residuos que contiene un triplete repetitivo de glicina (Gly)-X-Y, en donde X es prolina e Y son otros aminoácidos, frecuentemente hidroxiprolina; y un dominio no-colágeno (NCl) localizado en el carbono terminal (C-terminal) de cerca de 230 residuos. Cada cadena está codificada por un gen distinto (COL4A1-COL4A6), y se sitúan en pares en tres cromosomas diferentes. Las cadenas α1 y α2 (IV) son codificadas por los genes COL4A1 y COL4A2, respectivamente, en el cromosoma 13.
Los genes COL4A3 y COL4A4 codifican las cadenas α3 y α4 (IV), respectivamente, localizados en el cromosoma 2. Las cadenas α5 y α6 (IV) son codificadas por los genes COL4A5 y COL4A6 en el brazo largo del cromosoma X. Los cinco exones localizados en el extremo 3' de cada gen, codifican para el dominio NCl de la proteína y la mayoría de los exones restantes codifican para la porción colágena. Los extremos 5' de cada par de genes están contiguos, separados por secuencias de tamańo diverso que participan en la regulación transcripciona2,3.
Las seis cadenas de colágeno tipo IV forman aparentemente sólo tres moléculas determinadas de triple hélice llamadas protómeros, los cuales son designados como α1. α1, α2 (IV), α3. α4. α5 (IV) y α5. α5. α6 (IV). Estos protómeros crean cadenas de colágeno a través de uniones entre dos dominios NC1 de cada trímero para formar hexámeros y de uniones entre cuatro 7S dominios para formar tetrámeros con otros protómeros. Sólo tres grupos de hexámeros establecidos forman cadenas: α1. α1. α2 (IV)-α1. α1. α2 (IV), α3. α4. α5 (IV)-α3. α4. α5 (IV) y α1. α1. α2 (IV)-α5. α5. α6 (IV). El ensamblado de las cadenas de colágeno tipo IV es regulado por el desarrollo. La cadena α1. α1. α2 (IV)-α1. α1. α2 (IV) es un componente de todas las membranas basales de la "phyla" animal, mientras que las cadenas α3. α4. α5 (IV)α3. α4. α5 (IV) y α1. α1. α2 (IV)-α5. α5. α6 (IV) tienen una distribución restringida a los tejidos mamíferos. La cadena α3. α4. α5 (IV) se encuentra en el rińón (en la MBG y en algunas membranas tubulares), pulmón, testículo, cóclea y ojo; y la cadena α5. α5. α6 (IV) se encuentra en piel, músculo liso, esófago y en la cápsula de Bowman en rińón4.
Durante el desarrollo embrionario de la MBG del humano, la cadena α1. α1. α2 (IV) aparece al inicio temprano de la formación de las asas capilares, para ser reemplazada de manera gradual por la cadena α3. α4. α5 (IV) en el capilar glomerular ya maduro o por la cadena α5. α5. α6 (IV) en la cápsula de Bowman. Las diversas mutaciones y modos de transmisión del SA son responsables de la heterogeneidad en las características clínicas4.
Función del colágeno tipo IV
Las membranas basales son una estructura acelular que provee sostén a las células epiteliales y sirve para la compartamentalización de los tejidos, también influye en la proliferación, adhesión, migración y diferenciación de las células, contribuye a la polarización de los componentes subcelulares y la localización de receptores celulares y transportadores. Es un reservorio de factores de crecimiento, enzimas y proteínas plasmáticas.
La composición de las membranas basales varía de un tejido a otro y en un mismo tejido hay diferencia según la etapa de desarrollo y durante las fases de remodelamiento. Se componen de varias glucoproteínas de diversos tamańos que forman una compleja red. En el caso de la membrana basal glomerular (MBG), juega un papel importante en la filtración glomerular. Los principales componentes de todas las membranas basales son el colágeno tipo IV, laminina, nidógeno-elastina y proteoglicanos.
Tanto el colágeno tipo IV como la laminina forman cadenas entre ellas mismas, y estas redes se conectan con nidógeno-elastina y, a su vez, fijan otros componentes como proteoglicanos y fibulinas. Las diferencias funcionales en las diversas membranas basales se deben a la diferente composición de los diversos tipos de colágeno tipo IV y lamininas, así como de los constituyentes menores4.
Manifestaciones clínicas1-6
Afectación renal
La hematuria macro o microscópica es el síntoma más importante del SA. En los varones afectados hay hematuria microscópica persistente que comienza en etapas tempranas de la vida y pueden tener episodios de hematuria macroscópica precedidos por un cuadro de infección de vías respiratorias altas; la duración de los períodos de hematuria macroscópica varía entre 1 y 10 días, aunque en raras ocasiones puede persistir durante meses. La hematuria microscópica puede ser intermitente en mujeres portadoras de SALX y nińos pequeńos.
En los casos de Alport autosómico recesivo la hematuria es persistente tanto en hombres como en mujeres. La proteinuria es un hallazgo frecuente en los varones, y se incrementa con la edad; en raras ocasiones los pacientes pueden presentar síndrome nefrótico (edema, proteinuria, hipercolesterolemia, hipoalbuminemia). Las mujeres afectadas generalmente no tienen proteinuria, pero en el caso de estar presente, ésta puede ser leve o intermitente. Puede haber hipertensión arterial y su incidencia aumenta con la edad y la gravedad de la nefropatía.
La evolución depende del género y de factores genéticos. Los varones con SALX progresan a insuficiencia renal crónica terminal y generalmente la velocidad de progresión es similar entre los varones afectados de la misma familia, aunque se han reportado algunas familias con gran variabilidad en la progresión de la insuficiencia renal. Las mujeres portadoras de SALX generalmente tienen un curso benigno, pero pueden evolucionar lentamente a insuficiencia renal crónica terminal. La presencia de hematuria macroscópica en la nińez y síndrome nefrótico, así como la asociación de sordera neurosensorial y lentícono son factores de mal pronóstico en las mujeres afectadas. En los casos autosómicos recesivos, hombres y mujeres llegan a uremia en la segunda década de la vida.
Trastornos en la audición
El SA puede asociarse a sordera neurosensorial bilateral afectando aproximadamente al 55% de los varones y 45% de las mujeres. La pérdida auditiva nunca es congénita y suele ser paralela a la enfermedad renal. En la forma ligada al cromosoma X los varones afectados generalmente la manifiestan antes de los 10 ańos, inicialmente como disminución en la sensibilidad a tonos entre 2 000-8 000 Hz y el déficit va progresando a otras frecuencias. En las mujeres portadoras, el defecto auditivo puede ser detectado sólo por audiometría.
Trastornos oculares
Se han descripto trastornos oculares en el lente, retina y córnea en 15 a 30% de los pacientes con SA. El lentícono anterior es una protrusión en el aspecto anterior del lente por una acumulación anormal de colágeno. Este no está presente al nacimiento pero aparece en la segunda o tercera décadas de la vida, se asocia al síndrome de Alport en el 90% de los casos. También se han reportado cataratas subcapsulares como hallazgo frecuente en SA, así como miopía, anomalías en la pigmentación de la retina que se observan como granulaciones blancas o amarillentas que rodean la fóvea, erosiones corneales recurrentes y agujeros maculares.
Leiomiomatosis
Existe una forma de SA ligado al cromosoma X, en donde hay deleciones de los extremos 5' de COL4A5 y COL4A6. Estos paciente se caracterizan por presentar además de nefritis, sordera y afección ocular leiomiomatosis del esófago que puede ocasionar dolor retroesternal o epigástrico, disfagia, vómito postprandial y/o leiomiomatosis en el árbol traqueobronquial que se manifiesta con bronquitis recurrente, tos, disnea y estridor. Las mujeres afectadas pueden tener leiomiomas genitales e hipertrofia del clítoris.
REFERENCIAS
- 1.- Pirson Y. Making the diagnosis of Alport's syndrome. Kidney Int 1999;56:760-775
2.- Kashtan CE. Alport syndrome: abnormalities of type IV collagen genes and proteins. Ren Fail 2000;22:737-749
3.- Kashtan CE. Alport syndrome and the X chromosome: implications of a diagnosis of Alport syndrome in females. Nephrol Dial Transplant 2007;22:1499-1505
4.- Sindrome de Alport y nefropatía del colágeno IV (alfa3/alfa4). Nefrologia, Suplemento Extraordinario pre 2011. Mar.10904
5.- Gross O, Weber M, Fries JW, Muller GA. Living donor kidney transplantation from relatives with mild urinary abnormalities in Alport syndrome: long-term risk, benefit and outcome. Nephrol Dial Transplant 2009;24:1626-1630.
6.- Byrne MC, Budisavljevic MN, Fan Z, Self SE, Ploth DW. Renal transplant in patients with Alport's syndrome. Am J Kidney Dis 2002;39:769-675.
CORRESPONDENCIA:
Dra. Natalia Zabala
Sanatorio de la Trinidad Palermo.
Buenos Aires
Argentina
Email: natalialzabala @ hotmail.com
Comentario del revisor Carlos G. Musso, MD. PhD. Profesor Asociado de Fisiología Humana. Instituto Universitario del HIBA. Jefe de Fisiología Clínica Renal y de la Unidad de Diálisis Peritoneal. Hospital Italiano de Buenos Aires (HIBA). Argentina
Cabe seńalar que debido a que el síndrome de Alport puede cursar con macrohematuria en el contexto de infecciones respiratorias agudas, debe siempre tenerse presente su diagnóstico diferencial junto al de otras entidades como la nefropatía por IGA.
Comentario del revisor Prof. Juan F. Macías-Núńez, MD. PhD. Servicio de Nefrología. Hospital Universitario de Salamanca. Espańa
Dada la alteración que los pacientes con síndrome de Alport poseen en su colágeno tipo IV, y el consiguiente debilitamiento de su membrana basal glomerular, se ha postulado que el control de su hipertensión tanto arterial como intraglomerular (por ejemplo mediante el uso de inhibidores de la enzima convertidora, etc.) podría mitigar la progresión de su nefropatía hereditaria.