Indice del volumen Volume index
Comité Editorial Editorial Board
Comité Científico Scientific Committee
ESCLEROSIS TUBEROSA Y AFECCIÓN RENAL: REPORTE DE UN CASO CLÍNICO
Mariana Ciocchini1, Sebastián Lescano2, Andrés Rebaudi3,
Eduardo Rodriguez4, Ricardo Heguilén5
1DAOMI, Centro Integral de Nefrología, Institución afiliada a la Facultad de Medicina de la Universidad Nacional de Buenos Aires, Argentina
2Resonancia Magnética, 3Unidad de Urología, 4Unidad de Dermatología y 5Unidad de Nefrología
Hospital Juan A. Fernández. Buenos Aires, Argentina
Email: marianaciocchini @ gmail.com
Rev Electron Biomed / Electron J Biomed 2016;2:31-36.
RESUMEN
Una mujer de 27 ańos fue derivada a nefrología por poliuria, presentaba antecedentes de quistes renales y no tenía compromiso neurológico. Se realizó el diagnóstico de certeza de esclerosis tuberosa en la adultez a partir de dos criterios clínicos mayores conforme al "International Tuberous Sclerosis Complex Consensus Conference" de 2012: angiomiolipomas renales y angiofibromas faciales.
La Esclerosis tuberosa es una enfermedad multisistémica, de herencia autosómica dominante con una penetrancia casi completa siendo "de novo" en la mayoría de los casos y pudiendo manifestarse de muy diversas maneras aun dentro de un mismo linaje. La morbimortalidad en los adultos está dominada por el cuadro renal seguida del respiratorio.
Este caso ilustra sobre el alto nivel de sospecha que se debe tener ante pacientes con lesiones compatibles con esta enfermedad y la importancia de la colaboración entre los especialistas para alcanzar su diagnóstico.
PALABRAS CLAVE: Esclerosis tuberosa; Angiomiolipoma; Enfermedades renales quísticas; Angiofibroma; Hamartoma; Enfermedades pulmonares intersticiales.
SUMMARY:
A 27 year-old woman was initially referred to the division of nephrology because of polyuria and the finding of renal cysts. She did not have nervous system manifestations. According to the "2012 International Tuberous Sclerosis Complex Consensus Conference" the diagnosis of Tuberous Sclerosis Complex (TSC) was completed.
TSC is an autosomal dominant inherited multisystem disease with nearly complete penetrance. Clinical manifestations can vary widely even in patients belonging to a same linage. The morbidity and mortality depends on renal or respiratory complications on adulthood.
This report highlight the need of a high level of suspicions in patients with manifestations compatible with this disease and shows the importance of a multidisciplinary team working together to arrive to the appropriate diagnosis.
KEY WORDS: Tuberous Sclerosis; Angiomyolipoma; Kidney Diseases, Cystic; Angiofibroma; Hamartoma; Lung Diseases, Interstitial.
INTRODUCCIÓN
La esclerosis tuberosa, o enfermedad de Bourneville, descripta en 1862 por von Recklinhausen, es una enfermedad multisistémica, autosómica dominante, producida por muta-ciones en el gen "tuberous sclerosis complex 1" (TSC1) localizado en el brazo largo del cromosoma 9 (9q34) que codifica la hamartina o en el gen TSC2 ubicado en el brazo corto del cromosoma 16 (16p13.3) que codifica la tuberina1; dos tercios de las mutaciones son "de no-vo" con una penetrancia cercana al 100%2. La incidencia es de 1/6.000 -10.000 neonatos vi-vos y la prevalencia es de 1/20.0001. La pérdida del complejo hamartina-tuberina produce la activación constitutiva del mammalian target of rapamicyn complex (mTORC); por ello, los fármacos inhibidores del mTORC han surgido como alternativas terapéuticas3-9.
En 2012, se realizó la "International Tuberous Sclerosis Complex Consensus Conferen-ce" que actualizó los criterios diagnósticos y el manejo clínico de los pacientes con esclerosis tuberosa (TSC). Los criterios diagnósticos pueden ser genéticos o clínicos, dividiéndose éstos últimos en mayores y menores. El diagnóstico de certeza se realiza con el criterio genético, o con dos criterios clínicos mayores, o con un criterio clínico mayor y dos menores; en tanto que, con un criterio clínico mayor y otro menor, o con dos criterios clínicos menores, el dia-gnóstico es solo probable.
El criterio genético implica una mutación patogénica del gen TSC1 o del gen TSC2, la cual no se detecta en el 10 al 15 % de los pacientes y, justamente por ello, su ausencia no ex-cluye el diagnóstico de TSC (bajo valor predictivo negativo).
Los criterios clínicos mayores son:
1) Máculas hipomelanóticas (>3 de al menos 5 mm);
2) Angiofibromas (> 3) o placa cefálica fibrosa;
3) Fibromas ungueales (> 2);
4) Shagreen patch (placas elongadas, generalmente en localización dorso lumbar, con aspecto de piel de naranja);
5) Hamartomas retinianos (> 2);
6) Displasia cortical (sistema nervioso central);
7) Nódulos subependimarios;
8) Astrocitoma de células gigantes subependimario;
9) Rabdomioma cardíaco;
10) Linfangioleiomiomatosis pulmonar (LAM);
11) Angiomiolipomas renales o hepáticos (>2).
Mientras que los criterios clínicos menores comprenden:
1) Lesiones en "confetti"
2) "Pits" en el esmalte dentario (>3);
3) Fibromas orales (>2);
4) Parches arómicos retinianos,
5) Quistes renales múltiples;
6) Hamartomas no renales1.
CASO CLÍNICO
Paciente de sexo femenino de 27 ańos, argentina, oriunda de la provincia de Buenos Aires, que realizó una consulta nefrológica por poliuria con antecedentes de quistes renales diagnosticados cinco ańos atrás. Por ser contacto de tuberculosis (TBC) estaba recibiendo quimioprofilaxis con isoniazida (indicada en otra institución). Los antecedentes familiares relevantes incluían: madre (58 ańos) con ecografía renovesical normal; tía materna (61 ańos) con quistes y angiomiolipomas renales; tío materno (65 ańos) nefrectomizado en diálisis; abuela materna (fallecida) con cuadro pulmonar obstructivo; padre (64 ańos) con TBC pul-monar de reciente diagnóstico en tratamiento.
Los laboratorios de sangre y orina de 24 horas solamente evidenciaron poliuria acuosa y ferropenia absoluta; el sedimento urinario no presentó alteraciones; la ecografía renovesical mostró rińones de tamańo normal con quistes bilaterales y lesiones hiperecogénocas compati-bles con angiomiolipomas, la resonancia magnética (RM) de abdomen y pelvis con gadolinio (Gd) puso de manifiesto rińones de tamańo normal con angiomiolipomas renales bilaterales destacándose el de mayor tamańo de 21x17 mm en el rińón derecho, quistes renales simples y un quiste Bosniak tipo IIF en el polo inferior del rińón izquierdo, junto con tres quistes hepáticos simples (Figura 1). La prueba de restricción hídrica ratificó la sospecha de poliuria secundaria a polidipsia primaria en los meses estivales asociada a polaquiuria secundaria a uretritis.
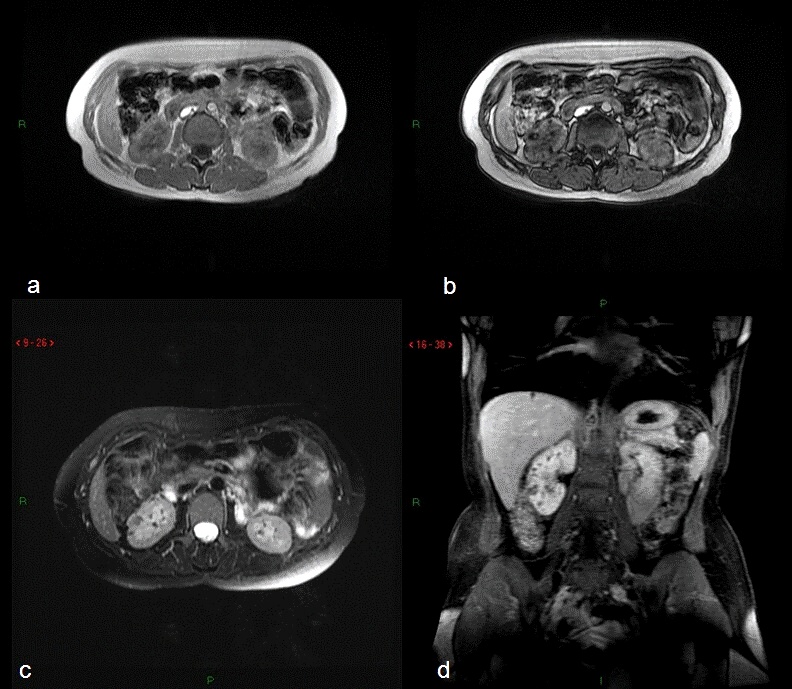
Figura 1.- Angiomiolipomas con imagen dominante en el rińón derecho,
a) Secuencia T1 en fase muestra imagen dominante con seńal espontaneamente interintensa.
b) Secuencia T1 fuera de fase evidencia seńal hipointensa por contenido graso. c) Secuencia T2 con supresión de la grasa muestra cancelación de la seńal que indica contenido de grasa macroscópica en la lesión.
d) Secuencia T1 con contraste endovenoso, muestra ausencia de captación de gadolíneo.
La evaluación dermatológica1,4 reveló más de tres angiofibromas faciales confirmados con biopsia cutánea (Figura 2.a).
La tomografía computada de tórax sin contraste endovenoso evidenció un patrón nodu-lillar difuso pulmonar (Figura 2.b) compatible con hiperplasia neumocística micronodular multifocal (MMPH) confirmada luego de descartar TBC por fibrobroncoscopía con lavado broncoalveolar (dado el antecedente de contacto de TBC) con test de la marcha normal1,4.
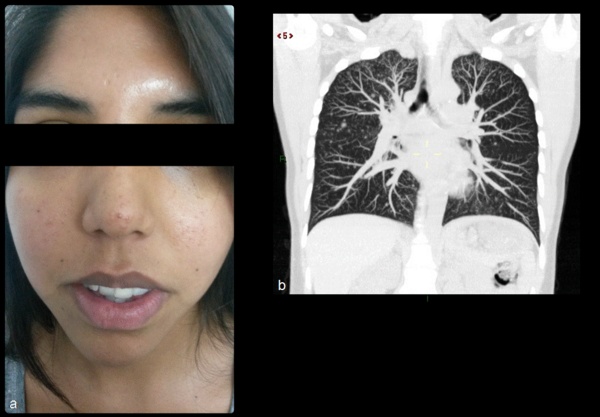
Figura 2.- Angiofibromas faciales. Tomografía de tórax con reconstrucción coronal,
muestra múltiples imágenes nodulares, de contornos bien definidos, de tamańo variable,
aunque menores a 10 mm, con distribución difusa en ambos campos pulmonares
con predominio de los lóbulos superiores.
Desde la perspectiva neurológica1,4, la paciente era políglota, carecía de antecedentes neuropsiquiátricos y la RM de sistema nervioso central con Gd fue normal.
A nivel ocular1,4, se hallaron un hamartoma retininiano en el ojo izquierdo que no presentó cambios en los controles sucesivos, descartando su posible origen infeccioso, y una cicatriz en la retina derecha. No presentó alteraciones cardiológicas1,4, ni arritmias en el electro-cardiograma ni rabdomiomas en el ecocardiograma trasntorácico. El examen odontológico1,4 mostró hipoplasia dentaria y gingivitis. No se encontraron hamartomas en otras localizaciones1,4.
Se realizó diagnóstico de certeza de TSC con dos criterios clínicos mayores: angiomiolipomas renales (> 2) y angiofibromas (> 3).
Por los angiomiolipomas renales, se le propuso a la paciente realizar tratamiento con everolimius, el cual rechazó. El quiste Bosniak tipo IIF no presentó cambios en los controles sucesivos. Se le contraindicaron los anticonceptivos hormonales, ya que los hamartomas son tumores derivados de las células epiteloides perivasculares (PEComas) con receptores hormonales4; se le indicaron medidas de nefroprotección, se le explicó el riesgo de transmitir la en-fermedad a su descendencia y la importancia de estudiar a sus familiares.
DISCUSIÓN:
Se presenta el caso de una paciente con diagnóstico de certeza de TSC efectuado en la adultez a partir de un cuadro nefrológico. Por los antecedentes familiares recabados, se sospecha que se trata de una mutación heredada por línea materna que habría comprometido, al menos, a dos generaciones (tíos maternos con cuadro renal y abuela materna con cuadro respiratorio), a diferencia de la mayoría de los casos que obedecen a mutaciones "de novo"2.
En contraste con los nińos en quienes la morbimortalidad está dominada por el cuadro neurológico, en los adultos con TSC, ésta depende en primer lugar del compromiso renal se-guido del pulmonar4.
A nivel renal, los pacientes con TSC pueden presentar angiomiolipomas múltiples, con su variante epitelioide, quistes renales, carcinoma de células claras, oncocitomas, linfangio-leiomiomatosis y glomeruloesclerosis focal y segmentaria secundaria1,3,4,9. Los angiomiolipomas renales pueden causar insuficiencia renal, hipertensión arterial, dolor y hemorragias retroperitoneales con compromiso vital por ruptura de los aneurismas que se desarrollan debi-do al componente epitelial de éstos3-6.
En el 50% de los pacientes se asocian angiomiolipomas y quistes renales lo que se con-sidera virtualmente patognomónico de TSC3 (como en nuestra paciente); de éstos, alrededor de 30 casos reportados a nivel mundial10, se deben al síndrome de genes contiguos TSC2-PKD1 (policystic kidney disease type 1) consecuencia de la deleción de los genes TSC2 y PKD1 ubicados adyacentemente en el brazo corto del cromosoma 16, con dańo renal severo y precoz, llegando a la adultez con rińones aumentados de tamańo y deformados, y con deterio-ro de la función renal11-13 (lo que no fue el caso de nuestra paciente).
En los pacientes con TSC, la principal lesión pulmonar es la LAM, la cual es un criterio clínico mayor, mas si se la encuentra asociada a angiomiolipomas renales, ambos deben con-siderarse como un único criterio dado que un tercio de los pacientes con LAM esporádica tienen angiomiolipomas renales. A nivel pulmonar también pueden encontrarse la MMPH (como en nuestra paciente) y el tumor de células claras1,4.
El uso de everolimus (un inhibidor del mTORC) a dosis de 10 mg/día por vía oral como tratamiento de los angiomiolipomas renales en pacientes con TSC que no presentan criterio quirúrgico está avalado por revisiones sistemáticas y un ensayo clínicos controlado aleatori-zados doble ciego4-6.
CONCLUSIÓN:
La TSC tiene una presentación mucho más variable de lo que se sospechaba hasta hace algunos ańos aun dentro de un mismo linaje. Desde la década del ochenta se sabe que, si bien, sigue siendo una enfermedad de las consideradas raras, su prevalencia es mayor a la estimada inicialmente, a partir de que se reconociera la existencia de casos con cuadros menos floridos o de distinta presentación, lo cual se ha visto favorecido por los consensos internacionales de 1998 y 20121,4.
Este caso resalta la importancia del alto índice de sospecha que deben tener los distintos especialistas ante pacientes adultos con lesiones compatibles con esta enfermedad, así como la importancia de la buena comunicación entre las distintas especialidades para poder alcanzar un diagnóstico de certeza.
Conflicto de Interes: todos los autores declaran que no tienen conflictos de intereses.
REFERENCIAS
-
1. Northrup H, Krueger DA, International Tuberous Sclerosis Complex Consensus Group. Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012 Iinternational Tuberous Sclerosis Complex Consensus Conference. Pediatr Neu-rol. 2013;49:243-54.
2. Northrup H, Koenig MK, Pearson DA, Au K. Tuberous sclerosis comlpex. En: Gene Review. NCBI Bookshelf.2015. [Acceso 5/2/2016].
3. Torres VE, Grantham JJ. Cystic Diseases of the Kidney. En: Brenner & Rector' The Kidney. Philadelphia: Elsevier. 2012:1646-7.
4. Krueger DA, Northrup H, International Tuberous Sclerosis Complex Consensus Group. Tuberous sclerosis complex surveillance and management: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol.2013;49:255-65.
5. Bissler JJ, Kingswood JC, Radzikowska E, Zonnenberg BA, Frost M, Belousova E, et al. Everolimus for angiomyolipoma associated with tuberous sclerosis complex or sporadic lymphangioleiomyomatosis (EXIST-2): a multicentre, randomised, double-blind, placebo-controlled trial. Lancet.2013;381:817-24.
6. Peng ZF, Yang L, Wang TT, Han P, Liu ZH, Wei Q. Efficacy and safety of sirolimus for renal angiomyolipoma in patients with tuberous sclerosis complex or sporadic lymphangioleiomyomatosis: a systematic review. J Urol 2014;5:1424-30.
7. Henske EP, Rasooly R, Siroky B, Bissler J. Tuberous sclerosis complex, mTOR, and the kidney: report of an NIDDK-sponsored workshop. Am J Physiol Renal Physiol 2014;306:F279-83
8. Malinowska IA, Lee N, Kumar V, Thiele EA, Franz DN, Ashwal S, et al. Similar trends in serum VEGF-D levels and kidney angiomyolipoma responses with longer duration sirolimus treatment in adults with tuberous sclerosis. PLoS One 2013;8:e56199.
9. Liang S, Salas T, Gencaslan E, Li B, Habib SL. Tuberin-deficiency downregulates N-cadherin and upregulates vimentin in kidney tumor of TSC patients. Oncotarget 2014;5:6936-46.
10. Autosomal dominant polycystic kidney disease type 1 with tuberous sclerosis. In: http://www.orpha.net. [Acceso 31/1/2016].
11. Brook-Carter PT, Peral B, Ward CJ, Thompson P, Hughes J, Maheshwar MM, et al. Deletion of the TSC2 and PKD1 genes associated with severe infantile polycystic kid-ney disease: a contiguous gene syndrome. Nat Genet 1994;8:328-32.
12. Sampson JR, Maheshwar MM, Aspinwall R, Thompson P, Cheadle JP, Ravine D, et al. Renal cystic disease in tuberous sclerosis: role of the polycystic kidney disease 1 gene. Am J Hum Genet.1997;61:843-51.
13. Martignoni G, Bonetti F, Pea M, Tardanico R, Brunelli M, Eble JN. Renal disease in adults with TSC2/PKD1 contiguous gene syndrome. Am J Surg Pathol 2002;26:198-205.
CORRESPONDENCIA:
Mariana Ciocchini
Montańeses 2731, 4to B,
Belgrano, CP 1428, CABA
Argentina
Email: marianaciocchini @ gmail.com