Indice del volumen Volume index
Comitť Editorial Editorial Board
Comitť CientŪfico Scientific Committee
- i) Epileptic activity is the result of uncontrolled insertion of substances into the cell membrane leading to an increased lateral pressure in the membrane and by this a mechanical damage to different channels, which leads to PDS.
- ii) Glial cells play a role in this context since they make "housekeeping" and when they cannot do this (e. g. destroyed by Pronase) epileptogenicity increases28.
- iii) The pacemaker neuron when epileptically active liberates intracellular substances from its dendritic surface on to neighboring neurons which are unspecifically activated.
- iv) Neuronal synchronization follows the epileptic depolarization and not (as is generally believed) neurons synchronize and from this generate PDs. This might be the basis for an epileptic focus.
- How can the duration of individual action potentials be increased on the background of an increase of membrane resistance?
- Given a parsimonious mathematical model for pacemaker potential, is it possible to increase its amplitude via manipulating membrane input resistance?
- Which mechanisms is the cell membrane equipped with, in order to fight membrane pollution, and which consequences are expected from weakening or invigorating such mechanisms?
- How does the ketogenic diet influence the physicochemical properties of the lipid bilayer? Is there an alternative antiepileptic diet that can be proposed on the basis of the membrane-pollution hypothesis?
- Are electroshock-induced seizures related to the release of membrane-polluting amphiphilic substances? What about insulin shock-induced seizures?
- Can the membrane pollution hypothesis provide a rational explanation to the wealth of epilepsy-associated genes that do not fit into the mainstream excitation-inhibition misbalance idea?
- Is it possible to find an isolate vertebrate neuron capable of producing PDS?
BASIC EPILEPSY MECHANISMS AND ALTRUP'S MEMBRANE POLLUTION HYPOTHESIS.
Suria Valdťs GarcŪa1, Josť Luis HernŠndez CŠceres2
1Facultad de Ciencias Mťdicas "Diez de Octubre", Universidad de Ciencias Mťdicas de La Habana.
2Centro de Neurociencias de Cuba.
La Habana. Cuba
Email: cacerjlh @ yahoo.com
Rev Electron Biomed / Electron J Biomed 2018;1:30-48
Comment of the reviewer Guillermo Videla MD. Profesor Adjunto de Neurofisiologia. Instituto Universitario del HIBA. Jefe de Secciůn de Trastornos de Marcha y Equilibrio. Servicio de NeurologŪa Hospital Italiano de Buenos Aires. Argentina
Comment of the reviewer Dra. Fiorella Martin Bertuzzi. Unidad de Estimulaciůn Magnťtica Transcraneana (UnEMaT). Neurůloga del Hospital Italiano de Buenos Aires. Argentina.
RESUMEN:
Se revisan las evidencias que apoyan la "Hipůtesis de Contaminaciůn de Membrana" (HCM) propuesta por Ulrich Altrup (1943-2007).
Altrup centrů sus estudios en los mecanismos celulares de la principal manifestaciůn electrofisiolůgica de la actividad epilťptica-las Despolarizaciones ParoxŪsticas (DPs). Sus estudios mostraron que: i) aun cuando la epilepsia se manifiesta en una intrincada red de neuronas y glŪas, una DP puede inducirse en una neurona aislada; ii) las DPs se acompaŮan de un aumento en la resistencia de membrana; iii) las DPs no son potenciales post sinŠpticos excitatorios gigantes, sino potenciales marcapasos gigantes; iv) la maquinaria sinŠptica no necesariamente estŠ afectada durante la epileptogťnesis, pero las entradas sinŠpticas a las neuronas marcapasos epileptizadas pueden modular el desarrollo de las DPs.
Respecto a los mecanismos bŠsicos, la HCM asume que: i) la actividad epilťptica resulta de la inserciůn descontrolada de sustancias anfifŪlicas en la membrana celular, conduciendo a un incremento en la presiůn lateral que daŮa mecŠnicamente diferentes canales iůnicos, conduciendo a DPs; ii) las cťlulas gliales desempeŮan una funciůn de "descontaminaciůn"; iii) las neuronas epilťpticamente activas liberan sustancias intracelulares desde sus dendritas activando inespecŪficamente las neuronas circundantes; iv) la sincronizaciůn neuronal aparece despuťs de la despolarizaciůn epilťptica y pudiera ser la base de un foco epilťptico.
La HCM contraviene algunos puntos de vista generalmente aceptados que conciben la epilepsia como un fenůmeno a nivel de red neuronal con la DP como un potencial postsinŠptico gigante. Al mismo tiempo, es capaz de explicar la mayorŪa de los hechos conocidos acerca del desarrollo de la epilepsia y concuerda con las opiniones recientemente expresadas respecto a las insuficiencias de la concepciůn generalmente aceptada de la epilepsia como un desbalance excitaciůn/inhibiciůn. Lamentablemente, esta hipůtesis es poco conocida entre los investigadores.
PALABRAS CLAVE:
SUMMARY:
Evidences supporting the "Membrane Pollution Hypothesis" (MPH) for epileptogenesis -proposed by Ulrich Altrup (1943-2007)- are reviewed.
Altrup centered his research on cellular mechanisms for the electrophysiological hallmark of epileptic activity: Paroxysmal Depolarization Shifts (PDS). His research led to the following observations: i) Even when epilepsy takes part in an intricate neuronal-glial network, a PDS can be elicited in an isolated neuron; ii) PDS is accompanied by membrane resistance increase; iii) PDSs are not gigantic excitatory postsynaptic potentials, instead, they are gigantic pacemaker potentials; iv) Synaptic machinery not necessarily is modified during epileptogenesis, but synaptic inputs to epileptized pacemaker neurons might modulate PDS development.
Regarding basic mechanisms, the MPH assumes that: i) Epileptic activity results from uncontrolled insertion of amphiphilic substances into the cell membrane leading to an increased lateral pressure in the membrane and thus to mechanical damage to different ionic channels, which leads to PDSs; ii) Glial cells play a "housekeeping" role in this process; iii) The pacemaker neuron when epileptically active releases intracellular substances from its dendritic surface on to neighboring neurons which are unspecifically activated; iv) Neuronal synchronization follows the epileptic depolarization and this might be the basis for an epileptic focus.
MPH counters commonly held views about epilepsy as a network phenomenon with the PDS as a gigantic postsynaptic potential. At the same time, it can explain most known facts about epilepsy development and it is congruent with recently expressed views about drawbacks in the usually accepted hypothesis of epilepsy as an excitation/inhibition misbalance. Unfortunately this hypothesis is rather unknown among researchers.
KEY WORDS:
INTRODUCCI”N
About 0.5% of humankind suffers from epilepsy1. Fortunately, available treatments are capable of keeping most of persons with epilepsy free from seizures, but still little is known about the basic mechanisms involved in epileptogenesis.
Theories about epilepsy are as old as philosophical thought. Worth of notice is the treatise about Epilepsy by Hippocrates (c. 460 BC-c. 370 BC) who denied any supernatural origin to the disease2:
-
"It appears to me to be nowise more divine nor more sacred than other diseases, but has a natural cause from where it originates, like other affections."Ö"The brain is the cause of this affection"Ö"In this disease as in all others, [the doctor] must strive not to feed the disease, but endeavor to wear it out by administering whatever is most opposed to each disease, and not that which favors and is allied to itÖwithout minding purifications, spells, and all other illiberal practices of a like kind."
It seems obvious that for understanding epilepsy, the basic functioning of the brain needs to be apprehended, and the way for it was paved only after the advent of the scientific method during the so called Scientific Revolution of the 17th Century. Still, the main heroes of that revolution were quite "naive" in their ideas about brain function. Thus Descartes, the genial creator of the orthogonal coordinate system, did not consider it necessary to make any observation or experimentation to discover how does the brain work, he simply knew how the brain must work. This passage from "Passions of the Soul" (1649), illustrates how the imagination of a genius can distort reality3:
- "Öand that this new blood that has just been formed from the alimentary juice, being driven to the heart with greater force than the blood from other parts of the body, enters the heart in greater abundance and produces a stronger heat there because it is coarser than the blood that has been rarefied many times in passing repeatedly through the heart. This makes it send to the brain spirits with unusually coarse and agitated parts; and these spirits, by strengthening the impression formed by the first thought of the loved object, compel the soul to dwell on that thought. This is what the passion of love consists in."
Newton, "the greatest genius of all times" developed a speculative scenario for nerve conduction. In "Opticks" (1704), Newton stated4:
- "ÖVibrations, being propagated along the solid Fibres of the optick Nerves into the Brain, cause the Sense of seeing. For because dense Bodies conserve their Heat a long time, and the densest Bodies conserve their Heat the longest, the Vibrations of their parts are of a lasting nature, and therefore may be propagated along solid Fibres of uniform dense Matter to a great distance, for conveying into the Brain the impressions made upon all the Organs of SenseÖI suppose that the Capillamenta of the Nerves are each of them solid and uniform, that the vibrating Motion of the ∆thereal Medium may be propagated along them from one end to the other uniformly, and without interruption".
On the other hand, Isaac Newton suggested that the speed of propagation of nerve activity is very high (like that of light5).
These views, expressed by the greatest knights of the scientific realm, influenced, and perhaps hindered, the advance toward a better understanding of nervous function.
Thus, when Hermann Helmholtz tried in 1850 to measure the speed of nervous impulse, he found himself discouraged by those who claimed that a process moving at the speed of light and manifested at the centimeter range, lays in the nano-second time scale and it would be practically impossible to detect using the technology available in middle of 19th century.
Helmholtz decided to challenge the infallible Newton, and found experimentally that the velocity of a nerve impulse is 27 m/s, (surprisingly agreeing with modern estimations). This value is even one order of magnitude lower than the speed of sound. This implied, that nerve impulse conduction was based on an unknown for that moment mechanism5.
Second part of 19th century marked important advances in clinical description and cataloguing of epilepsy modalities, but little was clarified regarding putative neurophysiological bases, besides Huggins Jacksonīs view that epilepsy "represents a sudden, excessive, disordered and recurrent discharge of cortical neurons"
Advances in understanding the bio-physical bases of nerve conduction allowed the proposal of physiologically sound hypotheses about epileptogenesis6.
In particular, thanks to a better understanding of synaptic mechanisms as well as the important role of inhibitory processes in brain function, the hypothesis of epileptic activity as a misbalance between excitation and inhibition had loomed. Nowadays, among a great diversity of proposed mechanisms, the idea of excitation/inhibition misbalance appears as the first-choice hypothesis for epileptogenesis.
What follows is a brief summary of prevailing ideas about epilepsy mechanisms as suggested by Maya Entenza (2010) in his excellent treatise6:
- Epileptogenesis is the process whereby a normal structure becomes hyper-excitable up to spontaneously elicit an epileptic seizure.
The normal functioning of the brain can be viewed as the result of the interaction of a high number of neuronal circuits where millions of neurons are interwoven. Neuronal networks, both in cortex, in hippocampus and the thalamus arise from the interaction of excitatory and inhibitory neurons, acting in a shrewdly established arrangement. Such balance is kept because the circuits are organized in such a way that excitatory and inhibitory inputs develop collateral connections with local interneurons, whereby inhibitory circuits -both anterograde and retrograde-are established that locally limit the excitation level. This way of functioning warrantees that networks are self-regulated, since an increment or a reduction in excitation is accompanied by an increase or a decrease in inhibition. At the cellular level a fine balance is also kept between excitation and inhibition, and each individual neuron is endowed with calcium and sodium- dependent membrane currents, especially at the level of dendrites, whose goal is to potentiate low amplitude synaptic inputs. These depolarizing currents are countered by voltage-dependent potassium currents activated by calcium, thus controlling the membrane potential, which prevents an exaggerated depolarization of the nerve cell.
According to the prevailing conceptions, the basic problem of epilepsy consists in understanding how and why a sudden and transient impairment of brain activity is produced, triggered by the rhythmic and synchronous discharge of a neuronal population, the pathogenic substrate of the epileptic event. This abnormal and paroxysmal discharge is clinically expressed in different ways, originating a wide repertory of clinical manifestations of the epileptic seizures. It is important to determine in each case, what caused such misbalance.
Most of the hypotheses and models of mechanisms for epileptogenesis arise from animal experimental models, where the mechanisms of paroxysmal discharges appear at the hippocampus after the injection of GABA-antagonists.
Since 1960īs epileptologists are trying to understand cellular mechanisms for epilepsy. High amplitude intracellular depolarizations, known as paroxystic depolarization shifts (PDSs), are followed by a hyperpolarization whose EEG counterpart is the wave following a peak discharge. In most of the studied neuronal groups, the PDS corresponds to a huge excitatory post synaptic potential mediated by glutamate and/or aspartate. To the depolarization resulting from the excitatory post synaptic potential the activation of voltage dependent ionic currents is added, mainly carried by sodium and calcium. The hyperpolarization following afterwards is originated by GABA-dependent currents as well as by intrinsic currents from the neuron, mainly potassium and potassium-calcium dependent.
Epilepsies are characterized by the capability of certain neurons to produce (PDSs). PDSs are initiated with a depolarization of the neuron that reacts with a high-frequency burst of action potentials, accompanied by a sustained depolarization, usually followed by a hyperpolarization of the neuron, finally returning to the resting potential. The initial phase of this burst is attributed to the activation of sodium channels associated to Kainate- or AMPA-type glutamatergic receptors, this leads to a fast inflow of sodium ions, depolarizing the cell. The sustained depolarization and the high-frequency burst are attributed to the activation of NMDA receptors, which produce a slow influx of calcium ions, as well as voltage-dependent calcium currents. The following sustained hyperpolarization has a fast component associated to the activation of GABA-A receptor associated chlorine channels, and a slow component due to the activation of GABA-B receptor associated potassium channels; under normal conditions, these prevent the spread of the discharges into other brain areas.
Neuronal synchronization during inter-ictal spikes as well as during the seizure are due to the existence of recurrent excitatory connections. However, other mechanisms may also be involved, among them: i) Electric coupling between neurons; ii) Extracellular ions concentration shifts; iii) Diffuse release of neuro-modulators; iv) Ephaptic interactions. Among the inhibitory mechanisms that might be impaired during epileptogenesis, there are: i) Blocking by Mg++ of NMDA-receptor-associated channels; ii) Na,K-ATPase activation with subsequent calcium outflow and after-discharge hyperpolarization; iii) Clearance of excess potassium and neurotransmitters by glial cells.
Hyperactivity of the excitatory system might lead to calcium inflow, dysfunction of the mitochondrial chain, lowered ATP production, lipases- and proteases activation, fatty acids release (which are substrates to synthesis of prostaglandins, leucotrienes, free radicals, and others). The activation of fast response genes might lead to apoptosis.
The above described picture is the result of intense research in brain mechanisms, observations made on persons with epilepsy as well as animal model research by many generations of epilepsy researchers.
Nevertheless, almost every piece of this proposed scenario is a matter of deep debate among specialists. Thus, a long standing discussion is related to whether epileptic activity takes place at the level of one neuron or is it manifested at single neuron level. Thirty years ago, one of us, also defended the idea that the minimal physiological entity capable of producing epileptic activity is the cortical column7. Today, this position is still held by most authors, for example, as Goldberg and Coulter8 expressed in 2013, "Epilepsy is inextricably a circuit-level phenomenon and cannot be understood outside this context". As it will be shown later, this view might be not as solid as it appears at first glance.
Today, new hypotheses, involving newly described molecular protagonists are being proposed, however, most of them rely on the classic idea of epilepsy being caused by a misbalance between excitation and inhibition. Thus for La Sarge and Danzer9 "Altered excitatory/inhibitory balance in the brain, in this case favoring increased excitability via reduced inhibition, might be an important mechanism by which changes in mTOR signaling promote epileptogenesis".
Similarly, Lason et al. stated10: "Neurochemical approach strongly supports the electrophysiological findings that seizures can be generated from excessively enhanced excitatory processes Öor from hypoactivity of neuronal inhibition, hyperactivity of glutamatergic transmission functional disturbances of the ligand- or voltage-gated sodium and calcium channels, insufficient GABAA receptor-mediated neurotransmission and extracellular potassium currents".
Huberfeld et al. (2013) addressed the crucial question of why epileptic fits are so rare and unpredictable, and their approach to find an explanation is through a combination of synaptic excitatory/inhibitory balance and ion homeostasis mechanisms11.
At the cellular level, epilepsy studies were benefitted when the theoretical and methodological/instrumental approach introduced by the Cambridge School was applied to find what the neuronal electrophysiological hallmark of epileptic activity is, and which the underlying mechanisms are.
In particular, it was possible to simultaneously obtain an electrocorticogram and an intracellular recording of a pyramidal cell during an epileptic episode in a rat (Figure1).
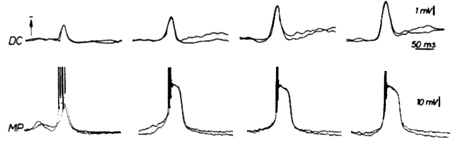
As it can be observed from Figure 1, epileptic activity in brainīs cortex is accompanied by high-amplitude and long-duration depolarizations. This is the event coined as "paroxysmal depolarization shift" (PDS).
It seems that understanding the nature of the PDS can be an excellent way to unravel epilepsy mechanisms, in a manner similar to the great boost basic neuroscience experienced after understanding the bases of the action potential. As noticed from the mainstream scenario shown above, most researchers view the PDS as a huge excitatory postsynaptic potential arising from misbalanced synchronic activity of neuronal masses.
The question about the nature of PDSs is also linked to the "epileptic neuron vs. epileptic neuronal network" controversy.
The prevailing opinion, till our days is that the PDS is merely a huge postsynaptic potential taking place in a reverberating pathological network of neurons. This conception fits very well into the widely accepted idea of epilepsy arising from a misbalance between excitation and inhibition.
Ulrich Altrup and Basic Epilepsy Mechanisms
Among the alternative hypotheses for epileptogenesis, the name of Ulrich Altrup (1943-2007), deserves a special place. Altrup was a full professor of physiology at the University of MŁnster, and worked during 35 years at the Institute of Physiology and the Institute for Experimental Epilepsy Research at the University of MŁnster, Germany. Referencess13-36 conform a sample of the papers published by him between 1979 and 2006.
Professor Altrup devoted most of his intellectual life to the study of epileptogenesis in the simplest known model for epilepsy: paroxysmal depolarizations shifts elicited in the neuron B3 from the buccal ganglia of the land snail Helix pomatia (Linnaeus, 1758), known under the common names of "orchards' snail", "vineyard snail", "Roman snail" or "Burgundy snail"- and probably one of the most known species of terrestrial mollusks.
Molluscan neurons have been a traditional model in neuroscience. In particular, studies by Erick Kandel of memory mechanisms on Aplysia californica were awarded with the Nobel Prize in 2000. More widely, invertebrate neurons have often proved useful in the study of basic mechanisms in nervous systems since they offer several decisive technical advantages when compared to vertebrate preparations. Among others are the existence of visually identifiable giant neurons which are well-known neuronal individuals. A further advantage is the structural and functional intactness of the ganglia kept under in-vitro conditions for many hours and even days.
The buccal neural ganglia of Helix contain four easily identifiable giant neurons, called B1, B2, B3, and B4. Among them, only B3 is capable of developing epileptic activity upon the application of proconvulsant substances (e. g. Pentylenetetrazol, Etomidate; Figure 2).
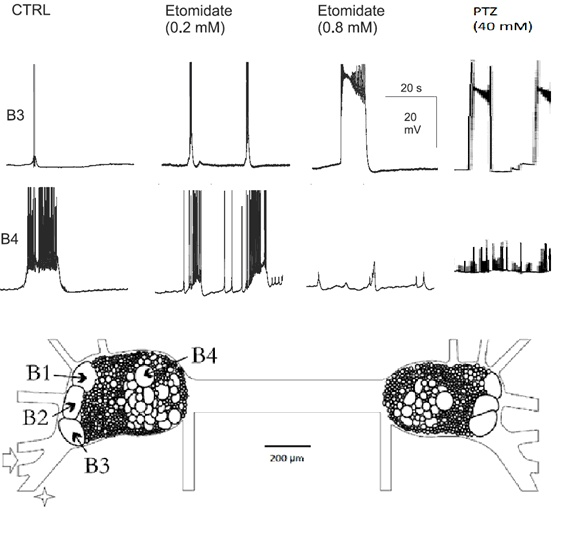
After systemic application of Etomidate (ETO) and Pentylenetetrazol (PTZ), mammalian as well as molluscan neurons generate epileptic paroxysmal depolarization shifts.
Both ETO and PTZ elicits PDs in a dose-dependent manner. Full PDSs appear at a ETO- and PTZ- concentrations of 0.8 mM and 40 mM respectively.
Altrup adhered to the working hypothesis that essential parts of the mechanisms underlying epileptic activity are constant irrespective of the considered type of human seizure and irrespective of the studied nervous system. Several observations demonstrate that the epileptiform activities of neurons in the buccal ganglia of Helix pomatia correspond to epileptiform activities recorded in vertebrate preparations and in the human nervous system. As an example, the buccal ganglia of Helix pomatia have been used to develop new antiepileptic substances and it was subsequently shown that the substances which proved antiepileptic in the invertebrate system were also antiepileptic in vertebrate nervous systems.
As expected, PDSs were found to be associated to a long-lasting inward current. However, unlike most of the classically described inward currents, PDS-associated inward current was accompanied by a paradoxical increase in membrane resistance (Figure 3). In particular, this counters to the mainstream idea that the depolarization during a PDS is due to a "fast inflow of sodium ions"
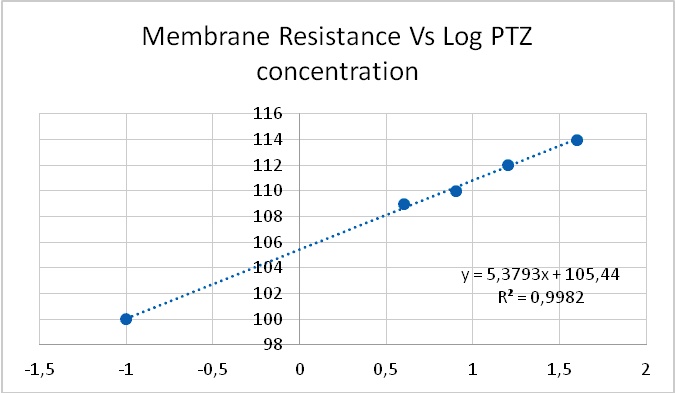
Indeed, the ionic mechanisms of this inward current were not clear37, even when it was initially postulated as being due to "a decreased potassium conductance."
At the same time, PDSs are paralleled by increases in the intracellular concentration of calcium (Figure 4).
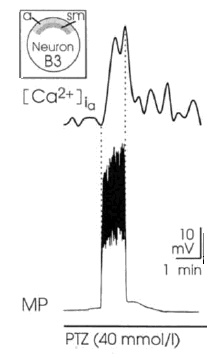
The possible synaptic nature of PDSs was ruled out through different means, in particular, PTZ is capable of eliciting PD in isolated Helix neurons38.
It was obtained that the effect of both PTZ and ETO upon postsynaptic potentials is completely uncorrelated with respect to their PDS-eliciting actions: ETO does not change the amplitude of excitatory post synaptic potentials whereas PTZ reduces them in a dose-dependent manner (Figure 6). Therefore, an explanation for epileptic actions of these two substances based on their effects upon postsynaptic potentials does not seem to be uncontroverted.
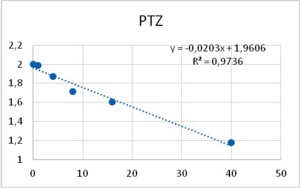
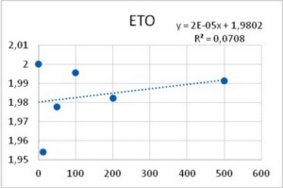
Figure 5. Reduction of Excitatory post synaptic potentials amplitude upon application of PTZ (left), and lack of any apparent effect from ETO (right). Abscissas: PTZ concentration in mM, and ETO concentration in ?M; ordinates: logarithm of percent change in PSP amplitude. Values were computed from data provided in36.
Among neurons B1-B4, only neuron B3 is capable to develop pacemaker potentials. Reports from other labs showed that PTZ is capable of inducing phase-shifting in circadian pacemaker neurons39. Altrup explored the effect of PTZ and ETO upon action potential parameters. It was found that PTZ increased the duration of action potentials in a dose-dependent manner (Figure 6).
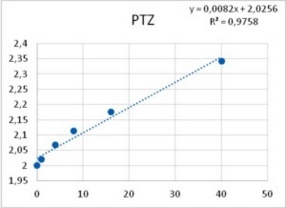
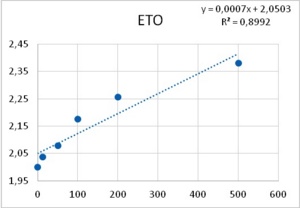
Figure 6. Increase in duration of action potentials upon the application of both PTZ (left), and ETO (right). Abscissas: PTZ concentration in mM, and ETO concentration in ?M; ordinates: logarithm of percent change in AP duration. Values were computed from data provided in36.
Further, Altrup studied the effect of convulsant substances upon pacemaker potentials. It was obtained that with the application of an epileptogenic drug, endogenous pacemaker potentials develop into PDSs. With increasing concentration of the drug, both the amplitude of pacemaker-depolarizations and delay of pacemaker-repolarization increased progressively finally resulting in PDSs. Only neurons which generated pacemaker potentials under control condition s could generate PDSs under epileptic conditions (Figure 7).
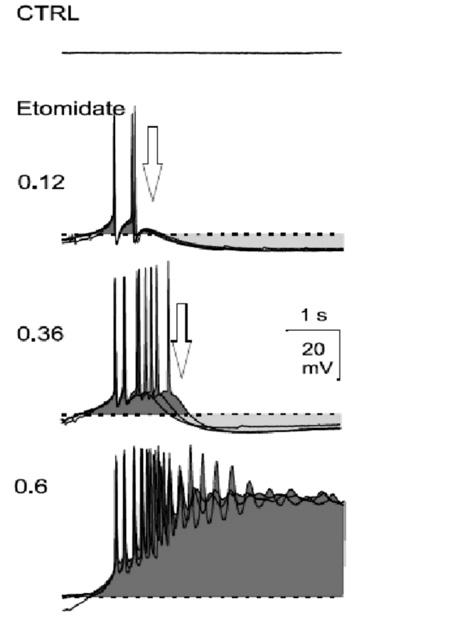
Synaptic input into neuron B3 could enhance or abolish the development of PDs. Specifically, synaptically induced potentials (induced via the stimulation of a nerve or from intra-ganglionic central pattern generators) did not participate in the generation of epileptiform activity but they can trigger both pacemaker potentials and PDSs.
Thus, the wealth of data collected by Altrup and his colleagues significantly modified prevailing ideas about epilepsy.
1. Even when epilepsy takes part in the intricate neuronal-glial network of the brain, the hallmark of epileptic activity -the PDS-can be elicited in an isolated neuron.
2. Unlike the common situation for excitatory processes, PDS are accompanied by an increase in membrane resistance.
3. PDSs are not gigantic excitatory postsynaptic potentials, as usually assumed, instead, they are gigantic pacemaker potentials.
4. Synaptic machinery not necessarily is modified during epileptogenesis. However, synaptic inputs to epileptized pacemaker neurons might exacerbate or diminish PDS development.
These facts, taken from the detailed study of epileptogenesis in a simple nervous system, on one hand, radically change some of the prevailing views about epilepsy mechanisms. At the same time, it can give a rational explanation to many known facts about epilepsy in humans.
The membrane pollution hypothesis
As it has been shown, processes -such as membrane resistance, action potential duration, pacemaker potential amplitude and duration- are altered by the tested epileptogenic drugs. Some processes showed a threshold concentration of 1 mM for PTZ and 12.5 µM for ETO. A 40-fold increase in concentration of both drugs induced PDSs, i.e., 40 mM of PTZ and 500 µM of ETO. The similarity in the dose dependency for several different neuronal mechanisms (leakage channels, voltage dependent potassium channels, pacemaker potentials to develop into PDS, membrane resistance) led Altrup to the idea that they resulted from an unspecific effect.
A possible explanation for such unspecific action could be through unspecific incorporation of epileptogenic substances into the cell membrane. To test this hypothesis, experiments were carried out on the air/water interface of a Wilhelmy film balance40. After forming the phospholipid monolayer on the saline subphase, the surface pressure in the layer was adjusted to 10 mN/m. At this pressure, all phospholipids molecules are in the liquid-condensed state and packed in a way that reasonably mimics the physical state of a biological membrane and still allows a certain rate of lateral diffusion.
Using a push-pull pump, the epileptogenic drugs were added to the saline beneath the phospholipid layer. During injection of the respective drug, pressure in the membrane rose exponentially. With an epileptogenic concentration of PTZ, the pressure increase was 6.9 ± 0.9 mN/m and the epileptogenic concentration of ETO increased pressure by 6.3 ± 0.9 mN/m36. Threshold effects of both drugs were 1/40th of their respective epileptogenic concentration. Dose-effect characteristics of incorporation in a phospholipid membrane thus corresponded to dose-effect characteristics of several different neuronal properties (Figure 8).
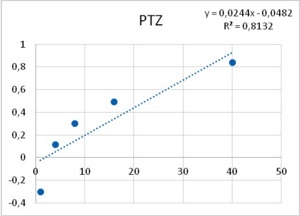
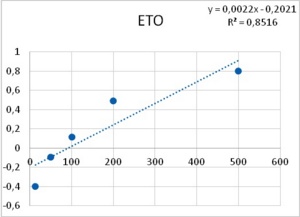
Figure 8. Increase in lateral pressure in an artificial membrane upon the application of both PTZ (left), and ETO (right). Abscissas: PTZ concentration in mM, and ETO concentration in µM; ordinates: logarithm of percent change in measured lateral pressure. Values were computed from data provided in36.
This body of results led Altrup to a revolutionary hypothesis for basic epilepsy mechanisms.
For Altrup, all these results together, did not directly show that epileptogenic drugs enter the neuronal membrane and that their incorporation increased membrane pressure and induced PDSs in neurons, which have expressed the membrane mechanisms of pacemaker potentials. At the same time, taking into account the bulk of observations here summarized strongly suggests that the key mechanism of epileptiform activity evoked by PTZ and Etomidate is their incorporation into the neuronal lipid bilayer, increasing membrane pressure and affecting conformational movements of membrane proteins. The amphiphilic drugs PTZ and Etomidate could attach to the external side of the membrane and then incorporate their lipophilic part into the lipid layer. This should increase membrane pressure and reduce membrane fluidity. Correspondingly, there were effects on membrane mechanisms in all studied neurons (B1, B2, B3, and B4). Epileptiform activity, however, was found in neuron B3 which can generate pacemaker potentials (Figure 9).
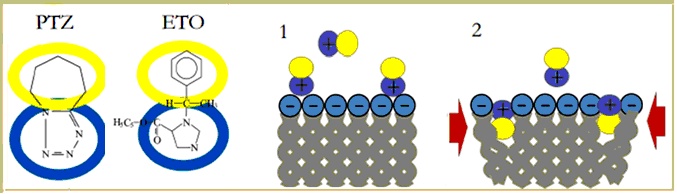
Figure 9. Summary of Altrupīs Membrane Polution Hypothesis for epileptogenesis. Amphiphilic drugs like PTZ and ETO (A) -or byproducts of abnormal brain cells metabolism- are postulated to adhere to the membrane surface (1) and incorporate the lipophilic part thus increasing membrane pressure (2, red arrows). Mechanical effects on the membrane pacemaker machinery are suggested to induce PDS development, and subsequently epileptic activity.
Summarizing, the key-points of Altrupīs membrane pollution Hypothesis are:
Aftermath
Professor Ulrich Altrup passed away in 2007, at the age of 63. It seemed that with him faded the idea of using mollusk neurons to study epilepsy mechanisms, and the membrane pollution hypothesis has not been yet quoted in scientific literature.
On one hand, ideas about epilepsy mechanisms continue to be centered on the assumption of network and synaptically based processes.
On the other hand, Altrup hypothesis has been capable of explaining the bulk of known facts about epilepsy, both clinical and in experimental, and -unlike mainstream views- can provide a reasonable explanation to some well-known facts as the paradox of the favorable effects of ketogenic diet41.
In recent years mounting evidence is accumulating that reveals the unsustainability of the traditional scenario and claim for the need of alternative hypotheses.
Thus Kaila et al42 concluded that "Changes in excitation-inhibition (E/I) balance are often used to explain epileptogenesis and seizure generation but, as should be obvious from the work reviewed above, the explanatory value of the E/I balance in the context of epilepsy is limited."
With the advent of Genomic Era, a group of genes associated to epilepsy has been identified. Paradoxically43, "The bulk of the mutations associated with epilepsy are not in inhibitory or excitatory ion channels, so the mechanisms by which mutations lead to seizures and epilepsy need to be broadened. It is possible that entirely new mechanisms of epilepsy will be discovered as a consequence of investigations into the cell biology of these newly-discovered gene defects."
Recently, the study of epilepsy mechanisms based on isolated snail neurons has been retaken38.
According to Erick Kandel44, a good hypothesis is the one that is capable of setting new, experimentally testable questions. In that sense, the membrane pollution hypothesis can pave the way for several questions, such as:
For now, the authors would like the scientific community to be aware of this nearly forgotten hypothesis that was promoted by a humble researcher who regarded that "We increasingly know more about smaller items even sub-quark particles or conditions, but this appears not to help with the fundamental problems of how the brain is working."
REFERENCIAS
-
1.- Ulrich A, Christian E, Markus R. Epilepsy Explained - A book for people who want to know more about epilepsy (Special edition for Epilepsy Action). Editorial: Medicine Explained. 2005. 368 p. ISBN 10: 3980963810 / ISBN 13: 9783980963817
2.- Hippocrates. On the Sacred Disease. Library of Alexandria (Publisher). 2007 ISBN 1465528040, 9781465528049
3.- Blom J. The Passions of the Soul. Descartes R. His moral philosophy and psychology. New York Univ Press. 1978. ISBN 0-8147-0999-0.
4.- Newton I. Opticks or a treatise of the reflections, refractions, inflections & colours of light. Cour Corp. 1952. P. 353
5.- Scott C. "Nerve Pulses and Reaction-Diffusion Systems. The Nonlinear Universe: Chaos, Emergence, Life (2007): 63-77.
6.- Maya C. Epilepsia. ECIMED. La Habana. 2010. 484 p. Available at: https://www.scribd.com/document/315905274/Epilepsia-Maya
7.- HernŠndez J L, Castellanos M, HernŠndez N. "Posible mecanismo de generaciůn de la espiga epilťptica". Neuroc de Cuba. 1978; 1: 36-42.
8.- Ethan M, Goldberg E, Douglas A. Mechanisms of epileptogenesis: a convergence on neural circuit dysfunction. Nat Rev Neurosci. 2013 May; 14(5): 337-349.
9.- LaSarge A, Candi L., Steve C. Mechanisms regulating neuronal excitability and seizure development following mTOR pathway hyperactivation. Front in Mol Neurosc (2014);18 (7):1-15.
10.- Wladyslaw L, Malgorzata C, Konrad R. Research advances in basic mechanisms of seizures and antiepileptic drug action. Pharmacol Rep 2013, 65: 787-801
11.- Gilles H, Le Duigou C, Le Van Quyen M, Navarro V, Michel Baulac M, Miles R. The Paradox of the Paroxysm: Can Seizure Precipitants Help Explain Human Ictogenesis? Neuroscientist. 2013; 19(5): 523-540
12.- Walden J, Straub H, and Speckmann E. Epileptogenesis: Contributions of calcium ions and antiepileptic calcium antagonists. Acta Neuro Scan. 1992; 86.S140: 41-46.
13.- Altrup U, Speckmann E, Caspers H. Axonal pathways and synaptic inputs of three identified neurons in the buccal ganglia of Helix pomatia. Malac. 1979: 473-476.
14.- Altrup U, Speckmann E. Responses of identified neurons in the buccal ganglia of Helix pomatia to stimulation of ganglionic nerves. Comp. Bioch. Physiol. 1982; 72: 643-657.
15.- Peters M, Altrup U. Motor organization in pharynx of Helix pomatia. J Neuroph. 1984; 52: 389-409.
16.- Altrup U. Inputs and outputs of giant neurons B1 and B2 in the buccal ganglia of Helix pomatia: an electrophysiological and morphological study. Brain Res. 1987; 414: 271-284.
17.- Altrup U, Speckmann E. Epileptic discharges induced by pentylenetetrazol: changes of shape of dendrites. Brain Res. 1988; 456: 401-405.
18.- Madeja M, Altrup U, Speckmann E. Synchronization of epileptic discharges: temporal coupling of paroxysmal depolarizations in the buccal ganglia of Helix pomatia. Comp Bio Physiol. 1989; 94C: 585-590.
19.- Altrup U, LehmenkŁhler A, Madeja M, Speckmann E. Morphology and function of the identified neuron B3 in the buccal ganglia of Helix pomatia. Comp Bio Physiol. 1990; 97A: 65-74.
20.- Altrup U, LehmenkŁhler A, Speckmann E. Effects of the hypnotic drug etomidate in a model nervous system (buccal ganglia, Helix pomatia). Comp Bio Physiol. 1991; 99C: 579-587.
21.- Haarmeier T, Altrup U, Speckmann J. Attenuation of a voltage-sensitive sodium current by GABA (identified neurons, buccal ganglia, Helix pomatia). Brain Res. 1994; 663 :131-639.
22.- Schulze-Bonhage A, Wiemann M, Altrup U, Wittkowski W, Speckmann E. Epileptic discharges induced by pentylenetetrazol: ultrastructural alterations in identified neurons and glial cells (Helix pomatia). Epile Res. 1995; 22 : 23-34.
23.- HernŠndez J, Altrup U, Speckmann E. Experimenten zu differenzirung switchen Epileptogenitat und epileptische Aktivitat . Epil -Blatter. Suppl. 1995; 26(8); 6
24.- Wiemann M, Wittkowski W, Altrup U, Speckmann E. Alterations of neuronal fibers after epileptic activity induced by pentylenetetrazol: fine structure investigated by calcium cytochemistry and neurobiotin labeling (buccal ganglia, Helix pomatia). Cell Tissue Res. 1996; 286: 43-53.
25.- Altrup, U, Peters M. Procedure of intracellular staining of neurons in the snail Helix pomatia. J Neurosci Methods. 1992; 5.(2): 161-165.
26.- Altrup, U, Haedder M, HernŠndez J, Malc Harek M, Meyer M, Galla H. Does epileptic activity follow membrane pollution with amphiphilic substances. Eur J Neurol. 2006; 13: 105-105.
27.- Wiemann M, Altrup U and Speckmann E. A method to study the effects of an epileptic focus on non-epileptic nervous tissue. J. Neur. Meth. 1996; 68 :137-41.
28.- Wiemann M, Jones D, Straub H, Altrup U, Speckmann, E. Simultaneous blockade of intracellular calcium increases and of neuronal epileptiform depolarizations by verapamil. Brain Res. 1996; 734:49-54.
29.- Altrup U, Hernandez J, Speckmann E. Vergleich zwischen zero-Mg2+- und Pentylentetrazol-induzierter epileptischer Aktivitšt in einem Modellnervensystem (Buccalganglien, Helix pomatia). Ep Blštt.1997;10 (27): 8
30.- Ulrich A. Informationstafeln Epilepsie (3rd Edition) BroschŁre, 41 Pages,Published in Nov Pharm. 2006. ISBN-13: 978-3-933185-66-2, ISBN: 3-933185-66-1
31.- Redecker C, Altrup U, Hoppe D, DŁsing R, Speckmann E. Effects of valproate derivatives. I. Antiepileptic efficacy of amides, structural analogs and esters. Neuroph. 2000; 39: 254-66.
32.- Redecker C, Altrup U, Hoppe D, Hense T, Kreie Ar, Rabe A et al. Effects of valproate derivatives. II. Antiepileptic efficacy in relation to chemical structures of valproate sugar esters. Neuropharm. 2000; 39: 267-81.
33.- Altrup U, Hšder M, Storz, U. Endogenous pacemaker potentials develop into paroxysmal depolarization shifts (PDS) with application of an epileptogenic drug. Brain Res. 2003; 1975: 73-84.
34.- Altrup U, ‹re A, Joschko A. Continuous Increase of Epileptogenic Effects Following Application of Proteolytic Enzymes (Buccal Ganglia of Helix Pomatia). Acta Biol Hung. 2004; 55 (1-4):269-72.
35.- Altrup, U. Pacemaker potentials are the physiologic basis of epileptiform activity in the buccal ganglia of Helix pomatia. Acta Biol Hung. 2004; 55 (4)): 261-68.
36.- Altrup U, Hšde M, HernŠndez J, Malcharek S, Michaela M, Hans-Joachim G. Epileptogenic drugs in a model nervous system. Electrophysiological effects and incorporation into a phospholipid layer. Brain Res. 2006 ;1122: 65-77.
37.- Walden J, Speckmann E, Witte O. Membrane currents induced by pentylenetetrazol in identified neurons of Helix pomatia Brain Res. 1998; 473 (2):294-305.
38.- Brenes O, Carabelli V, Gosso S, Romero A, Carbone E, Montarolo P et al. Subconvulsant doses of pentylenetetrazol uncover the epileptic phenotype of cultured synapsin-deficient Helix serotonergic neurons in the absence of excitatory and inhibitory inputs. Epilep Res. 2016; 127,10.1016/ j.eplepsyres.09.008.
39.- Bir S, Khalsa S, Gene B. Phase-shifting of a neuronal circadian pacemaker in Bulla gouldiana by pentylenetetrazol. Comp Biochem Physiol C Toxicol Pharmacol. 1992; 101(3): 557-560
40.- Rhoderick E, Howard L. Using Monomolecular Films to Characterize Lipid Lateral Interactions. Meth Mol Biol. 2007 ; 398: 41-58.
41.- Kossoff E, McGrogan J. Worldwide use of the ketogenic diet. Epil. 2005; 46(2): 280-289
42.- Kai K, Ruusuvuori E, Seja P, Voipio J, Puskarjov M. GABA actions and ionic plasticity in epilepsy. Current Opinion in Neurobiology. 2014; 26:34-41
43.- Staley K. Molecular mechanisms of epilepsy. Nat Neur. 2015; 18(3): 367-372.
44.- Kandel E. Cellular Basis of Behavior: An Introduction to Behavioral Biology (Freeman, San Francisco, 1976).727 p.
CORRESPONDENCE:
Dr. Jose Luis HernŠndez CŠceres
Investigador Titular Centro de Neurociencias de Cuba
La Habana
Cuba
Email: cacerjlh @ yahoo.com
Comment of the reviewer Guillermo Videla MD.
Profesor Adjunto de Neurofisiologia. Instituto Universitario del HIBA. Jefe de Secciůn de Trastornos de Marcha y Equilibrio. Servicio de NeurologŪa
Hospital Italiano de Buenos Aires. Argentina
Este artŪculo describe principalmente la hŪpotesis de contaminaciůn de membrana de Ultrich Altrup como mecanismo epileptogťnico y la confronta con lo que hoy estŠ mŠs aceptado.
En la actualidad se piensa que la epilepsia se origina por un desbalance a nivel de los circuitos neuronales entre la actividad excitatoria e inhibitoria. En condiciones fisiolůgicas, un incremento en el nivel de la excitaciůn de las redes neuronales desencadena mecanismos de autorregulaciůn aumentando la actividad inhibitoria de otras redes con las que se interconecta. Las investigaciones a nivel celular se han focalizado en determinar las neuronas capaces de generar paroxismos de despolarizaciůn, que no son otra cosa que grandes ondas de despolarizaciůn seguidas de una hiperpolarizaciůn reactiva. Aunque la aceptaciůn mŠs universal es que el mecanismo epileptogťnico es a nivel de una red neuronal y no intracelular.
Altrup mediante sus investigaciones desarrolla su "hipůtesis de contaminaciůn de membrana", en la cual postula que ciertas sustancias actuarŪan a nivel interno de la membrana celular provocando una modificaciůn en su resistencia y generando cambios a nivel de la presiůn intracelular lateral, lo que finalmente llevarŪa a una afectaciůn de los canales iůnicos ubicados en ella y generarŪan asŪ una onda despolarizante.
En mi opiniůn personal ambas teorŪas pueden complementarse y no son necesariamente excluyentes, es probable que en los průximos aŮos y con las actuales lŪneas de investigaciůn encontremos una nueva teorŪa mŠs general y unificadora.
Comment of the reviewer Dra. Fiorella Martin Bertuzzi . Unidad de Estimulaciůn Magnťtica Transcraneana (UnEMaT). Neurůloga del Hospital Italiano de Buenos Aires. Argentina.
La epileptogťnesis es un tema de fundamental interťs para los neurůlogos. Las convulsiones son uno de los trastornos mŠs impactantes en nuestra prŠctica y en el imaginario social. Desde la antigŁedad y la edad media, en donde a las crisis se le atribuŪa un carŠcter divino o demonŪaco, estos eventos paroxŪsticos resultan fascinantes para quienes estudian el campo de la salud y su origen es territorio de grandes controversias.
Como bien revelan los autores, existen mķltiples explicaciones, que se basan en la premisa del desequilibrio entre influencias excitatorias e inhibitorias, ya sea por exceso de las primeras o por falla de las segundas. La "hipůtesis de la contaminaciůn de membrana", de Ulrich Altrup, explica varios mecanismos celulares que podrŪan estar involucrados en el inicio de la actividad epilťptica a nivel neuronal.