Indice del volumen Volume index
Comité Editorial Editorial Board
Comité Científico Scientific Committee
- Edad: Es sin lugar a duda el factor de riesgo más importante, como ya se mencionó anteriormente, la prevalencia aumenta significativamente al aumentar la edad. 21
- Antecedentes familiares de EA: Aproximadamente el 20% de los pacientes con diagnóstico de EA tiene dos o más familiares de primer grado afectados; el patrón de herencia es por lo general autosómico dominante.
Se han identificado que varias mutaciones familiares principalmente con inicio de la enfermedad en la quinta o sexta década de la vida involucran el gen APP, se piensa que estas mutaciones generan metabolismo anormal de la proteína
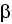 amiloide, llevando esto a niveles aumentados de la misma, de manera crónica7.
amiloide, llevando esto a niveles aumentados de la misma, de manera crónica7.
Otras mutaciones también se han encontrado en EA de inicio temprano, localizadas en el gen PS-1 (Presenilina-1) ubicado en el cromosoma 14 y en el de la PS-2 (Presenilina-2) en el cromosoma 1. Las mutaciones de la PS-2 se han encontrado en pocas familias, mientras que de la PS-1 se han encontrado más de 160 mutaciones diferentes7-8
- Apolipoproteina E (ApoE): El polimorfismo
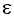 4 de esta lipoproteína constituye el factor de riesgo genético de mayor relevancia.
4 de esta lipoproteína constituye el factor de riesgo genético de mayor relevancia.
La ApoE es uno de los mayores transportadores de colesterol en sangre periférica; en el líquido cerebroespinal, es la principal proteína de transporte lipídico y también realiza función de transportador de
amiloide.
Se conocen 3 polimorfismos:
2, 3 y 4, siendo el alelo 2 muy poco común, mientras que el 3 es el más común; el polimorfismo 4 se encuentra entre el 15% y el20% de la población, las personas homocigóticas para este polimorfismo tienen 50% de riesgo para generar EA antes de los 60 ańos y los heterocigóticos tienen un 50% de riesgo a los 70 ańos.
Los ratones que expresan ApoE
4 muestra desinhibición del mecanismo de seńalización de la ciclofilina A en los pericitos de las venas cerebrales, resultando en un dańo en las mismas, disminución de la barrera hematoencefálica y neurodegeneración independiente de la proteína amiloide7,9-10. Una vez la cascada fisiopatológica destructiva del tejido neuronal ha iniciado, la ApoE no influye en su progresión.
- Género: Las mujeres están en mayor riesgo, aun cuando se ajusta a su sobrevida más larga, antes se pensaba que esta predisposición era principalmente en post menopaúsicas, sin embargo, la Women´s Health Initiative Memory Study comprobó que la utilización de estrógenos aumentaba el riesgo de demencia11
- Nivel educativo: Varios estudios han asociado el mayor nivel educativo con reducción del riesgo de EA y presentación más tardía de los síntomas. Se han realizado hipótesis de que el motivo es el mayor desarrollo de reserva cognitiva; sin embargo, el entrenamiento cognoscitivo y los ejercicios mentales no han demostrado beneficios importantes en el retraso del inicio de los síntomas, al contrario, la actividad física sí ha demostrado que disminuye la atrofia cerebral7.
- Factores de riesgo cardiovascular: Múltiples estudios han demostrado que el síndrome metabólico (Diabetes Mellitus en manejo con insulinas o no, hipercolesterolemia, hipertensión y obesidad) aumenta el riesgo de EA7, 12,13.
- Fragilidad: Cada vez es mayor la evidencia que asocia la fragilidad con EA. En un estudio con 823 pacientes, se evidenció que cada punto en las escalas de fragilidad (medida por la fuerza de agarre, índice de masa corporal, velocidad de la marcha y agotamiento) aumentaba el riesgo en un 9%. 30
- Preocupación en el propio individuo, en un informante que le conoce o en el clínico, porque ha habido un declive significativo en una función cognitiva, y
- Un deterioro sustancial del rendimiento cognitivo, preferentemente documentado por un test neuropsicológico estandarizado o, en su defecto, por otra evaluación clínica cuantitativa.
- Los pacientes que presentaron síntomas cognoscitivos tenían una mayor probabilidad de progresar a demencia.
- De los pacientes a quienes se les diagnosticó demencia, la mayoría presentaron síntomas depresivos al inicio.
- La aparición de la demencia es independiente de la presencia de quejas iniciales de dificultades cognoscitivas subjetivas.
- 1. La regulación glutamatérgica mejora la eficiencia de los neurotransmisores a traves de la transmisión de impulsos, lo que se supone, aminora los síntomas cognoscitivos.
- 2. La Memantina previene el exceso de entrada de calcio a las neuronas, siendo este mecanismo neuroprotector.
- Demencia multiinfarto: demencia debido al efecto acumulativo de múltiples infartos grandes, corticales o subcorticales, secundarios a oclusión aterotrombótica o embolica de arterias de mediano y gran calibre. La edad y la hipertensión arterial son los principales factores de riesgo. Describe curso clínico de inicio brusco y progresión escalonada.
- Demencia por infarto estratégico: demencias ocasionados por infarto único estratégico a nivel cortical y subcortical. En estructuras cerebrales: circunvolución angular, en la porción caudal de la rodilla de la capsula interna y los infartos talámicos paramedianos.
- Demencia de pequeńo vaso: ramas perforantes de las arterias cerebrales a nivel cortical y subcortical. Es una de las causas más frecuentes de demencia vascular. El cuadro clínico corresponde a una demencia subcortical: enlentecimiento psicomotor, queja de memoria, apatía, depresión, parkinsonismo, ataxia, incontinencia urinaria. Se distinguen 2 subtipos: a) demencia vascular subcortical: estado lacunar, enfermedad de Binswanger y angiopatía cerebral autosómica dominante con infartos subcorticales y leucoencefalopatía (CADASIL) y b) demencia vascular corticosubcortical: angiopatía hipertensiva, angiopatía amiloide, vasculopatía colágena con demencia.
- Demencia por hipoperfusión o isquémico- hipóxica: originada por infartos secundarios a hipotensión, bradiarritmias y estenosis arteriales graves. Las causas más frecuentes de esta demencia son el ataque global transitorio, hipoventilación pulmonar y alteraciones en el transporte de oxígeno al cerebro. Los infartos se pueden situar en los territorios de los grandes vasos, pero son especialmente sensibles las zonas fronterizas entre las zonas irrigadas por la arteria cerebral media y posterior
- Demencia hemorrágica: son consecuencia de lesiones residuales tras un proceso hemorrágico, las manifestaciones dependen de la zona lesionada. Las causas más frecuentes son la hipertensión arterial, aneurismas y malformaciones arteriovenosas y venosas.
- Con estado de ánimo deprimido: desde labilidad emocional hasta depresión mayor. Es el subtipo más frecuente y requiere tratamiento activo.
- Con ideas delirantes: puede además incluir cuadros psicóticos como alucinaciones y/o falsas identificaciones.
- No complicado: cuando no predomina ningún síntoma psicopatológico en especial.
- Con trastorno del comportamiento: agitación, deambulación errática, reacciones catastróficas y generan más impacto en su entorno que el deterioro cognitivo asociado.
- D: Demencia
- D: Delirium (fluctuación cognitiva)
- aV: alucinaciones Visuales
- P: Parkinsonismo
- Características centrales (esenciales para el diagnóstico de demencia de Cuerpos de Lewy probable o posible):
Demencia con declinación cognitiva progresiva o de suficiente magnitud para intervenir con la función social u ocupacional. La alteración de la memoria puede ocurrir no necesariamente en estadios tempranos, pero se desarrolla con la progresión. Déficits en los test de atención, función ejecutiva y en la habilidad visoespaciales son prominentes. - Características clave (2 son suficientes para un diagnostico probable. 1 realiza un diagnóstico posible):
- Fluctuacion cognitiva, variación prominente en el estado de alerta y en la atencion.
- Alucinaciones visuales recurrentes, tipicamente bien formadas y detalladas
- Manifestaciones espontaneas de parkinsonismo
- Caracteristicas sugestivas (si una o mas estan presentes junto con una o mas de las caracteristicas claves, el diagnostico es probable. Si las caracteristicas claves estan ausentes pero existe una o mas de las caracteristicas sugestivas,el diagnostico es posible):
- Comportamiento durante el sueńo con movimiento oculares rapidos
- Sensibilidad neuroleptica severa
- Imágenes SPECT SCAN o PET SCAN que revelan baja tasa de transporte o concentracion de Dopamina en elos ganglios basales.
- Carcateristicas de apoyo:
- Metaiodobenzilguanidina (MIBG) reducida en la escintigrafia miocardica
- Imágenes SPECT SCAN que revelan hipofusion occipital
- Estructuras del lo temporal medial relativamente conservadas en las imágenes TC/RMI
- Caídas recurrentes y sincopes
- Perdidas de la conciencia transitorias e inexplicables
- Disfuncion autonomica
- Otros tipos de alucionaciones
- Ilusiones sitematizadas
- Depresion
- Actividad de onda lenta prominente en el electroencefalograma, con ondas planas temporales transitorias
- Diagnostico menos probable:
- En presencia de infartos cerebrales evidentes con signos de focalizacion al examen clinico
- En presencia de otras patologias o desordenes cerebrales que llenen criterios en parte o talmente para su presentación clinica
- Si el parkinsonismo solo se presenta en estadio de demencia severa.
- Secuencia temporal de los sintomas
La demencia de Cuerpos de Lewy es diagnosticada cuando precede o es simultanea con el parkinsonismo. El término demencia de la enfermedad de Parkinson, debe usarse para describir la demencia que ocurre en presencia de una enfermedad de Parkinson bien establecida. Para realizar diagnosticos difenrenciales entre estas dos entidades, el curso de un ańo desde el inicio de los sintomas de parkinsonismo al desarrollo de la demencia cuenta como regla para diagnosticar demencia de Cuerpos de Lewy. - Deterioro cognoscitivo leve en enfermedad de Parkinson
- Edad avanzada
- Severidad de los sintomas extrapiramidales
- Mayor duracion de la enfermedad
- Sexo masculino
- Sintomas motores atipicos
- Desarrollo temprano de alucionaciones
- Criterios 2011 para variante comportamental:
Al menos 3 de los síntomas que indican deterioro progresivo del comportamiento y/o cognición: desinhibición temprana, apatía, perdida de la empatía o simpatía, comportamiento compulsivo, perseverante o estereotipado, hiperoralidad y cambios dietarios, perfil neuropsicológico que demuestre déficit de función ejecutiva con conservación de memoria y habilidades visoespaciales.El diagnóstico es posible cuando cumple los criterios anteriores; probable cuando se soportan por neuroimágen y definitivo cuando haya hallazgos histopatológicos7.
- Variante afasia primaria progresiva:
Al menos 1 de los 2 síntomas característicos: agramatismo, apraxia del discurso
Y al menos 2 características adicionales: comprensión alterada de frases sintácticamente complejas, pobre comprensión de palabras únicas y reconocimiento de objetos1. - Deterioro cognoscitivo:
Se recomienda el uso de inhibidores de la acetilcolinesterasa. Debe tenerse en cuenta que producen efectos gastrointestinales colaterales y raramente bradicardia, que pueden requerir limitaciones de las dosis.- Donezepil: Dosis de 5 mg por 4 semanas, luego 10 mg/día
- Rivastigmina oral: Dosis de 1,5 mg dos veces al día, incrementar 1.5mg cada 2 a 4 semanas hasta un máximo de 6 mg dos veces al día
- Rivastigmina transdermica: Dosis de 4.6 mg, por 24 horas por 4 semanas, luego incrementar a 9.5mg por 24 horas
- Galantamina: Dosis de 4 mg, 2 al día, incrementar 8 mg 2 veces al día a las 4 semanas, luego llevar a 12mg 2 veces al día en la semana 8.
Antagonistas del receptor N metil de Aspartato (NMDA)
- Memantina: Dosis de 5 mg por una semana, luego 5 dos veces al día, luego 10mg en la mańana y 5mg en la tarde por una semana, luego 10mg 2 veces al día.
- Enlentecimiento psicomotor
Se recomiendan inhibidores de la acetilcolinesterasa a las mismas dosis que ya fueron descritas.
Se recomienda el uso de agonistas dopa:
Carbidopa/levodopa: dosis de 25 mg a 100 mg 2 veces al día. Para tolerabilidad se pueden repartir 3 veces al día - Apatía
Inhibidores de la acetilcolinestarasa
Inhibidores selectivos de la recaptación de serotonina (ISRS): La experiencia es anecdótica. Medicamentos activadores como Sertralina y Bupropion pueden ser útiles, evitar antidepresivos tricíclicos por el efecto anticolinérgico.
Café: 1 o 2 tasas antes de las 2 pm - Psicosis: (alucinaciones - ilusiones)
Se recomiendan inhibidores de la acetilcolinesterasa a las mismas dosis que ya fueron descritas.
Quetiapina: dosis de 12.5 mg, 1 hora antes del inicio esperado de las alucinaciones. Se puede administrar a necesidad o con dosis de mantenimiento titulando gradualmente 12.5mg a 25mg, máximo 200mg al día o según aparición de prolongación del segmento Qt. Puede exacerbar parkinsonismo, por lo que se recomienda usar la dosis menor con la que se obtenga respuesta y a la menor duración.
Clozapina: dosis de 12.5 mg cada noche al dormir, incrementar 12.5 mg hasta un máximo 50mg 3 veces al día. Se debe vigilar posible desarrollo de discrasias sanguíneas. - Características motoras del parkinsonismo:
Se recomienda carbidopa/levodopa a dosis de 25mg a 100mg 2 veces al día,
Terapia física y ejercicio
Terapia ocupacional
Fonoaudiología - Depresión y ansiedad
Se recomienda
Escitalopram: dosis de 10mg a 20mg, de una forma titulada.
Venlafaxina: Dosis de 37.5 mg hasta 225mg.
Citalopram: dosis de 10mg a 20mg
Sertralina: dosis de 25mg hasta un máximo 200mg
En Ansiedad se puede ańadir Buspirone 5 mg 2 veces al día, incrementando hasta una dosis de 15 a 30 mg día - Insomnio
Melatonina: dosis de 1mg a 3 mg, 1 hora antes de dormir
Trazodona: dosis de 25 mg cada noche, hasta máximo 100 mg
Mirtazapina: dosis de 7.5mg a 15 mg, cada hasta un máximo de 45mg cada noche
>Quetiapina: dosis de 12.5mg - Somnolencia diurna:
Café 1 a 2 tazas antes de las 2 pm
Metilfenidato: dosis de 1 a 5 mg día hasta máximo 20 mg
Modafinil: dosis de 100mg cada mańana hasta 400 mg máximo - Disautonomía:
Se recomienda hidratación, actividades frecuentes que van desde levantarse con frecuencia, cruzar las piernas, disminución de la sal durante la dieta.
Fludrocortisona: dosis de 0.1mg hasta 0.2 mg al día
Midodrine: dosis de 5 mg 3 al día hasta máximo 10 mg - Incontinencia urinaria
Trospium: dosis de 20 mg cada noche
Darifeacin dosis de 7.5 mg - Constipación:
Control del uso de medicamentos anticolinérgicos
Incrementar actividad física mejorar hidratación
Dietas que contengan laxantes osmóticos y estimulantes
DEMENCIAS, II.
1Ángela María Benjumea Salgado MD, 2Sebastián López Velásquez MD,
2Laura María Cano Méndez MD.
1Medicina Interna y Geriatría y 2Cirugía, Universidad de Caldas.
Manizales, Caldas, Colombia.
Email: ambsco4 @ gmail.com
Rev Electron Biomed / Electron J Biomed 2018;3:22-43.-
Comentario de la revisora Dra. Cynthia Marińansky . Geriatra de planta de la Unidad de Geriatria del Hospital Durand de Buenos Aires Directora de la Especialización en Geriatria Universidad Maimonides.
Comentario de la revisora Dra Silvina Dahl. Geriatra. Hogar Ledor Vador, Buenos Aires. Argentina.
RESUMEN
La demencia es un síndrome adquirido, de naturaleza orgánica, que se caracteriza por un deterioro permanente de la memoria y de otras funciones cognitivas y frecuentemente se presenta acompańado de otras manifestaciones psicopatológicas. Su importancia radica en que se espera que el número de adultos con Alzheimer aumente en los próximos ańos y, al menos, un 50% para el 2030. Por lo tanto, los médicos nos veremos enfrentados a gran cantidad de pacientes con este complejo síndrome, lo que nos debe hacer encender las alarmas para realizar grandes esfuerzos en la terapéutica y para estar al tanto de los múltiples detalles del mismo.
En este capítulo describimos los diversos tipos de demencia, incluyendo la enfermedad de Alzheimer, demencia frontotemporal, demencia vascular y demencia por cuerpos de Lewy, además ofrecemos recomendaciones para un completo abordaje inicial e integral de los pacientes. Se revisan las diversas características, etiologías, presentaciones clínicas y terapéuticas.
PALABRAS CLAVE: Demencia. Alzheimer. Frontotemporal. Vascular. Cuerpos de Lewy.
ABSTRACT. DEMENTIA
Dementia is an acquired syndrome, of an organic nature, characterized by a permanent deterioration of memory and other cognitive functions and often accompanied by other psychopathological manifestations. Its importance is that it is expected that the number of adults with Alzheimer's will increase in the coming years and at least 50% by 2030. Therefore, physicians will be confronted with a large number of patients with this complex syndrome, which should make us turn on the alarms to make great efforts in therapeutics and to be aware of the multiple details of this syndrome.
In this chapter we describe the different types of dementia, including Alzheimer's disease, frontotemporal dementia, vascular dementia, body Lewy dementias, as well as recommendations for the initial and general approach of patients. The diverse characteristics, etiologies, clinical and therapeutic presentations are reviewed.
KEY WORDS: Dementia. Alzheimer. Frontotemporal. Vascular. Body Lewy dementias
INTRODUCCIÓN
La demencia es un síndrome adquirido, de naturaleza orgánica, que se caracteriza por un deterioro permanente de la memoria y de otras funciones cognitivas y frecuentemente se presenta acompańado de otras manifestaciones psicopatológicas; ocurre sin alteración del nivel de conciencia y afecta el funcionamiento social y ocupacional. La etimología del término nos lleva a su origen latino demencia = de (fuera de) + mens (mente) + ia (estado de).
La discusión fundamental acerca del concepto de demencia se centra en la necesidad de que exista o no un deterioro de la memoria para diagnosticar demencia, dado que en algunas formas de demencia la afectación de la memoria es tardía y la afectación de otras funciones cognitivas (lenguaje, capacidades visoespaciales, emocionalidad, personalidad y cognición: abstracción, cálculo, juicio, funciones ejecutivas) aparece antes de esta, por lo tanto se propone que bastaría tener compromiso de tres de ellas para hacer el diagnóstico de demencia. Se discute así mismo, si el síndrome demencial debe definirse como un déficit cognoscitivo global, en este sentido hay que seńalar que las demencias no afectan a todas las funciones cognitivas con igual gravedad, manifiestan predominantemente la alteración del funcionamiento de ciertas áreas cerebrales y no otros, lo que permite su clasificación y diferenciación clínicas.
Epidemiología
Existen al menos 30 estudios importantes sobre la prevalencia del síndrome demencial en distintos lugares del mundo, y sus resultados son muy dispares. Esta variabilidad se ha atribuido a factores metodológicos, aunque en los últimos ańos se ha avanzado en el proceso de homogeneización metodológica, con los que los resultados tienden a acercarse. En cuanto a los estudios internacionales, las cifras varían desde el 3,5% hasta el 28%. Además de la mayor edad, los factores de mayor peso a la hora de explicar las variaciones de prevalencia son: procedencia de la muestra (en cuanto a si se incluye o no pacientes ingresados en residencias) y los criterios de inclusión, así como el grado de deterioro cognitivo. Los estudios realizados en residencias de ancianos muestras tasas de prevalencias muy altas y alcanzan hasta el 78%, teniendo en cuenta que la demencia es la causa más frecuente de ingreso a hogares. Estudios de ancianos en la comunidad reportan prevalencias de 3,6% a 4,8%.
La prevalencia de demencia en personas mayores de 65 ańos es de 8%, cifra que puede doblarse si se incluye a personas con deterioro cognitivo leve o formas leves de demencia. La tasa de conversión de pacientes con demencia leve o DCL a demencia establecida es del 10% a 12% anual.
La forma de demencia más frecuente es la enfermedad de Alzheimer (EA), en Estados Unidos actualmente afecta a 5.2 millones de personas en su mayoría ancianos y la incidencia dobla cada 6 ańos después de los 50 ańos. La segunda causa de demencia después de la enfermedad de Alzheimer, con manifestaciones clínicas, neuropatológicas y neuroimágenes variables. Existe de hecho un interés incrementado en la relación entre lesión vascular y desarrollo de patogénesis y manifestaciones clínicas de EA. La demencia fronto temporal (DFT) es la tercera causa de demencia neurodegenerativa, después de EA y demencia por cuerpos de Lewy, es la demencia de comienzo temprano con reportes de rango de edad muy amplio desde 21 ańos hasta 89. Los datos epidemiológicos de la DFT son escasos, especialmente en la población anciana y los estimativos de prevalencia e incidencia varían ampliamente, esto último en parte a la heterogeneidad clínica de la demencia, a los criterios diagnósticos y a las regiones de estudios con mayor o menor contribución de causas genéticas.
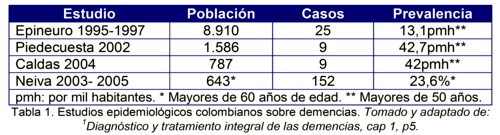
En Colombia se han realizado 4 estudios epidemiológicos de campo (Tabla 1), todos concuerdan en que se afectan más las mujeres y el subtipo de demencia más frecuente es la EA, seguida de la demencia vascular y también en nuestro país como en el resto del mundo la prevalencia de demencia aumenta con la edad.
Manifestaciones clínicas y diagnósticas
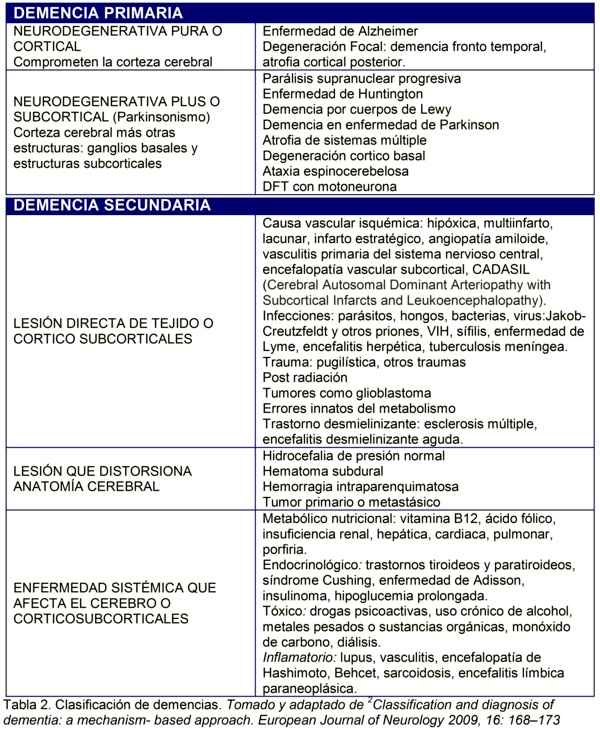
El síndrome demencial adquiere rasgos clínicos característicos tanto en cuanto al tipo de déficit neuropsicológico como a su presentación y evolución según la enfermedad causante subyacente; existen numerosas causas de demencia (Tabla 2).
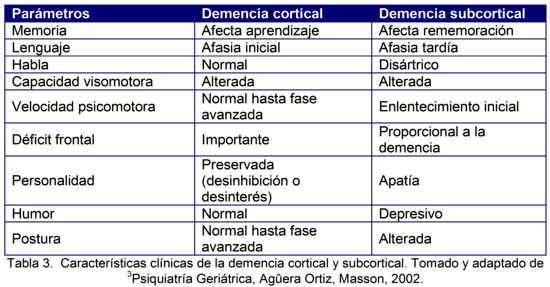
En cuanto al tipo de déficit neuropsicológico se diferencias dos subsíndromes de acuerdo con las estructuras neuroanatómicas más afectadas en cada caso: demencia cortical, demencia subcortical y cortico subcortical. Las características clínicas de los tipos de demencia se muestran en la tabla 3.
Diagnóstico
El diagnóstico de demencia es fundamentalmente clínico y debe seguir una serie de pasos a mencionar: historia clínica con anamnesis dirigida, exploración psicopatológica y neuropsicológica con pruebas de cribado y exploración complementaria.
El diagnóstico debe de incluir los siguientes aspectos:
- 1. Diagnóstico de la presencia de deterioro cognitivo
2. Caracterización clínica del deterioro cognitivo como demencia
3. Diagnóstico de presunción de la forma de demencia
4. Grado de repercusión funcional, social, familiar.
La queja de memoria o cognoscitiva debe ser tenida en cuenta y siempre evaluarse con la presencia de un familiar cercano ojalá su cuidador e indagar aspectos incluidos en el cuestionario de neurología para precisar áreas de afectación, los antecedentes familiares y personales relacionados como consumo de alcohol y otras sustancias psicoestimulantes, historia familiar de demencia, antecedentes psiquiátricos.
Es de anotar que el gold standard para diagnóstico de demencia es la batería neuropsicológica de lo cual no hay un instrumento estandarizado y validado, pero al menos debe evaluar: orientación global, memorias, pensamiento abstracto y capacidad de juicio, lenguaje oral y escrito, praxias, ejecución y control.
DEMENCIA TIPO ALZHEIMER
La enfermedad de Alzheimer (EA) es la causa más común de demencia a nivel mundial. Fue descrita por Alois Alzheimer en 1906 en una mujer de 51 ańos de edad con características claras de demencia4. Luego de su muerte le realizó autopsia al cerebro encontrando múltiples placas y ovillos característicos de la patología. Por 60 ańos se pensó que consistía en una entidad exótica, sin embargo, con el advenimiento de las nuevas tecnologías y la mayor comprensión de sus síntomas, signos y fisiopatología, la llevó al primer lugar en las demencias en los ancianos.
En diciembre del 2013 el G8 estableció que la demencia debe convertirse en una prioridad con la meta de encontrar la cura o un tratamiento que modifique la enfermedad para el 2025.
Epidemiología
Se estima que 40 millones de personas (la mayoría mayores de 60 ańos) tienen diagnóstico de demencia, un hecho que espera que se duplique cada 20 ańos, al menos hasta el 20505-6.
Se trata de una enfermedad principalmente de ancianos (mayores de 65 ańos), la prevalencia en menores de 50 ańos es menor a 1 en 4.000, siendo un 30% atribuido a la EA; para la edad de 75 ańos, la prevalencia es del 15% y para mayores de 85 ańos del 35% al 50% en Estados Unidos5-6.
En los últimos 5 ańos, múltiples estudios de cohortes se han realizado, en uno de ellos en el que la incidencia fue directamente comparada entre sub cohortes, la incidencia ajustada a la edad encontró que las personas que nacieron a partir del 2000 tienen 24% menos riesgo de desarrollar la patología que los de los 90; también compararon los factores de riesgo e imágenes cerebrales encontrando menor atrofia cerebral y dańo vascular, a pesar del incremento en los factores de riesgo cardiovascular presentes en esta población; estudios en Estados Unidos han hallado similares resultados5,6.
Esta estimación sobre el aumento de la prevalencia será en su mayoría dependiente de países en desarrollo, con población joven.
Factores de riesgo
Existen 7 factores de riesgo conocidos como "Los 7 grandes" que son: diabetes, obesidad, inactividad física o mental, depresión, tabaquismo, bajo nivel educativo y la dieta, el estudio Rotterdam demostró que la eliminación de estos factores redujo la incidencia en un 30%. Lo que evidencia el gran potencial que tiene la medicina preventiva, pero también la necesidad de estrategias terapéuticas para el 70% restante7, 14.
Fisiopatología
Existen actualmente miles de artículos sobre este tema, por lo que un abordaje completo requeriría un solo compendio y aun en esas circunstancias sería una tarea titánica. A continuación, expondremos las características más generales para el entendimiento de los procesos que conllevan (al menos los entendidos hasta el momento) a la enfermedad de Alzheimer.
Por 30 ańos se ha considerado que la acumulación anormal de la proteína amiloide (A) y las proteínas tau en placas y ovillos están asociados directamente con los procesos neurodegenerativos. Pero a través de los diferentes estudios y avances nos hemos dado cuenta de que es mucho más complejo que simplemente asumir la causalidad lineal de la hipótesis amiloide5.
Como se mencionó anteriormente, la mayor evidencia de que las proteínas tau y A causan neurodegeneración provienen de estudios en familias con Alzheimer donde se han encontrado mutaciones en la APP, PS-1 y PS-2. La APP es la precursora de los péptidos A y su mutación afecta la escisión y agregación; PS-1 y PS-2 componen la subunidad catalítica de las secretasas  , las cuales escinden la A42; las mutaciones en las PS causan alteraciones en el procesamiento de APP lo que conlleva a generación de péptidos A más largos y más hidrófobos5,8
, las cuales escinden la A42; las mutaciones en las PS causan alteraciones en el procesamiento de APP lo que conlleva a generación de péptidos A más largos y más hidrófobos5,8
Las proteínas Tau son a su vez requisito para el diagnóstico de EA, sin embargo, las mutaciones del gen tau causa demencia frontotemporal sin formación de placas, lo que indica que las proteínas tau actúan independiente de la A en la neurodegeneración. Es totalmente plausible que ambas proteínas actúen de manera sinérgica potenciando sus efectos tóxicos5.
Las conformaciones tóxicas de ambas proteínas que ocurren durante la enfermedad son llamadas "semillas", por su capacidad para inducir mayor acumulación de péptidos anormales contribuyendo a la diseminación de la enfermedad, sin embargo la naturaleza de estas conformaciones (oligomérica o fibrilar, modificaciones post translacionales) requiere mayores investigaciones; en el momento se conoce que la conformación oligomérica de A se une a varios receptores de membrana incluidos priones, pero la relevancia de esto aún no se ha aclarado5.
Por lo anteriormente mencionado, se pensaría que si la proteína A juega un papel central, al interrumpirse su producción se podría contrarrestar la enfermedad, este mismo pensamiento lo han tenido múltiples investigadores y ahora se conoce que la primera estación de escisión para la producción de A es mediada por la secretasa BACE1, altamente expresada en las neuronas y que hace parte de múltiples procesos biológicos, incluyendo la plasticidad neuronal, la inhibición de la BACE1 produce un procesamiento alternativo de la APP generando péptidos activos diferentes a A. Aún se están investigando los inhibidores de la secretasa. Las investigaciones sobre inhibidores de la secretasa (segunda fase), fueron interrumpidos por los múltiples efectos adversos5,15
Diagnóstico
Antes de adentrarnos en los criterios diagnósticos, es importante hablar del concepto: "Enfermedad de Alzheimer preclínica"16, The National Institute on Aging-Alzheimer´s Association (NIA-AA) desarrollo una clasificaciones para el abordaje preclínico, esta clasificación aplica para población en riesgo genético o por edad y se basa principalmente en biomarcadores (marcadores de A y marcadores de neurodegeneración (ND) incluyendo la medición de proteínas Tau en liquido cerebroespinal y patrones volumétricos de atrofia por resonancia magnética nuclear)17.
La NIA-AA estableció 3 estadios así: Estadio 1 A+/ND-; estadio 2 A+/ND+ y estadio 3 A+/ND+ con evidencia de alteraciones cognoscitivas o comportamentales sin cumplir criterios para deterioro cognoscitivo leve. Con fines investigativos, se ha creado el estadio 0 que consiste en A-/ND. La Clínica Mayo propuso incluir el concepto de "Sospecha de Patología Diferente a Alzheimer" (SPDA) que comprende los pacientes con biomarcadores o imágenes de neurodegeneración sin exceder los puntos de corte de Amiloidosis, curiosamente los pacientes en el subgrupo SPDA son en su mayoría hombres y mayores de 80 ańos17.
Las proporciones en cada estadio son: estadio 1: 10-15%, estadio 2: 15%, SPDA: 25%, el porcentaje restante se encuentran entre el estadio 0 y 3.
Si bien la NIA-AA tiene criterios diagnósticos de Demencia y en particular de tipo Alzheimer18, los criterios más difundidos son los del Diagnostic and Statistical Manual of Mental Disorders, fifth edition (DSM-5)19 que son los siguientes:
Criterios diagnósticos
A. Evidencias de un declive cognitivo significativo comparado con el nivel previo de rendimiento en uno o más dominios cognitivos (atención compleja, función ejecutiva, aprendizaje y memoria, lenguaje, habilidad perceptual motora o cognición social) basadas en:
B. Los déficits cognitivos interfieren con la autonomía del individuo en las actividades cotidianas (es decir, por lo menos necesita asistencia con las actividades instrumentales complejas de la vida diaria, como pagar facturas o cumplir los tratamientos).
C. Los déficits cognitivos no ocurren exclusivamente en el contexto de un delirium.
Dentro de la evaluación clínica, es importante tener en la cuenta que indagar directamente al paciente por su patología generalmente es infructuoso por las mismas características de la enfermedad, manifestando que no presenta alteraciones (anosognosia); por lo que contar con un familiar o cuidador es fundamental en la valoración, a él o ella se debe indagar por las actividades de la vida diaria tanto físicas como instrumentales y a la vez corroborar información dada por el paciente7.
Cómo ya sabemos la EA afecta principalmente la memoria episódica por lo que debemos interrogar a los familiares si los pacientes repiten constantemente las conversaciones, olvidan asistir a reuniones, leen el periódico o ven televisión pero recuerdan muy poco o nada de lo vieron o leyeron; en el transcurso de la patología también se pueden presentar alteraciones lingüísticas incluida la incapacidad de recordar los nombres de sus familiares o amigos, también presentan dificultades para encontrar las palabras a usar en una conversación fluida o viso espaciales encontrándose el paciente desorientado dentro del hogar o con dificultades para realizar viajes cortos20.
La Asociación Americana de familiares con Enfermedad de Alzheimer y Otras Demencias publicó signos de alarma sobre los problemas de memoria, son: disminución de la memoria reciente que afecta el desempeńo en el trabajo, dificultades en el desempeńo de tareas familiares, problemas del lenguaje, desorientación en tiempo y lugar, pobre o disminuida capacidad de juicio, problemas con el pensamiento abstracto, extraviar cosas, cambio del ánimo o del comportamiento, cambios en la personalidad y disminución de la iniciativa.
Los cambios comportamentales son muy comunes en estos pacientes a medida que la enfermedad progresa, generando gran carga para el cuidador. Por lo general la apatía ocurre en el inicio de la enfermedad generando perdida de la iniciativa y anhedonia. Se estima que el 30% de los pacientes en estadios iniciales presentan depresión, incluyendo abulia, hiporexia o anorexia y alteraciones del sueńo (el insomnio en estos pacientes puede ser causado por apneas del sueńo, mioclonus o como reacciones adversas de los medicamentos). Debido a la gran proporción de pacientes con depresión en el marco de la EA, a la carga que genera para el paciente y el cuidador, es mandatoria la realización de una tamización, la más utilizada es la escala de Yesavage, un cuestionario de 15 ítems que se califican como si o no, otorgándole acorde al ítem un valor de 0 o de 1, se considera que el punto de corte es 6, recordar que esta escala es de tamización y que una vez se pase el punto de corte deben aplicarse los criterios DSM-5 de depresión. También se encuentra la Escala de Depresión de Hamilton (HRSD) que tiene la ventaja de evaluar parámetros no cognitivos, además el analfabetismo y la desorganización no interfieren en la evaluación, por lo que se ha postulado como ideal en el paciente demente7.
Demencia y Depresión
Esta relación se sintetiza en 3 supuestos clínicos21:
1. Desarrollo de un episodio depresivo en un paciente con una demencia establecida:
Consiste en un reto diagnóstico, ya que los signos y síntomas (bradipsiquia, hiporexia, pérdida de peso, alteraciones del sueńo, hipoquinesia, aislamiento social, labilidad emocional y dificultades cognoscitivas) son compartidas entre las dos condiciones.
En las diferentes revisiones se ha esclarecido que la principal diferencia es la temporalidad, mientras la demencia requiere meses o ańos para sí instauración, la depresión por lo general es un proceso de semanas, es por esto (como ya hemos mencionado) que la buena relación médico-paciente- familiares contribuye a disminuir la cantidad de pacientes a quienes se les puede ofrecer ayuda.
2. Presencia en el curso de un episodio depresivo primario de quejas de fallos cognoscitivos que pueden confundirse con una demencia:
Es la entidad clínica conocida como pseudodemencia de la depresión, ya que produce alteraciones cognoscitivas, pero en estudios neuropsicológicos se ha aclarado que estas alteraciones son dependientes del componente atencional.
3. Demencia que debuta con un síndrome depresivo:
En los diferentes estudios en los que se ha comparado la respuesta a los antidepresivos por parte de pacientes con síntomas cognoscitivos o sin ellos, se demostró que:
La ansiedad (principalmente cuando se quedan solos en el hogar o cuando deben interactuar con personas desconocidas), la paranoia y las ideas delirantes son parte del espectro de la EA, la idea delirante más común consiste en que los vecinos, amigos o familiares le están robando.
El 75% de los pacientes hacen emergencias comportamentales caracterizadas por agresión física o verbal hacia alguno de los familiares o cuidadores. Pueden desarrollar inhibición y comportamientos inapropiados21. Para resumir, los pacientes con demencia presentan alteraciones en el rendimiento cognoscitivo (juicio y raciocinio, abstracción, calculo, atención y memoria), alteración conductual y afectiva (depresión apatía, desinhibición y agresividad) y alteraciones en la función ejecutiva (planificación, resolución de problemas, flexibilidad mental, multi-tarea y seguimiento de acciones).
El examen físico y principalmente el neurológico es fundamental no solo para diferenciar diferentes tipos de demencia sino para identificar causas reversibles de EA.
Comúnmente el examen neurológico es normal, sin embargo la presencia de déficit focales sugiere un origen vascular de la demencia, del 10% al 30% de los pacientes con EA pueden desarrollar signos de enfermedad de Parkinson (rigidez, bradiquinesia y temblor), las alteraciones en la marcha y el balance generalmente hacen pensar en demencia vascular, hidrocefalia de presión normal o parálisis supranuclear progresiva, aunque hay que aclarar que las alteraciones persistentes de la marcha pueden ocurrir en estadios más avanzados de la enfermedad21.
Es fundamental entender que una vez se hace el diagnostico de demencia indica un compromiso de sus capacidades que no retornará a un estado basal por lo que, antes de atrevernos a dar el diagnóstico, debemos excluir las causas reversibles de deterioro cognoscitivo, las cuales van del 35% al 40%, para esto es de gran utilidad la mnemotecnia DEMENTIA22 que consiste en:
D: Drugs (medicamentos): analgésicos, anticolinérgicos, antihipertensivos, antipsicóticos
E: Eyes and Ears: dificultades en la audición o visión conocido como deprivación sensorial
M: Metabolic (metabólicas): como hipo o hiperglucemia, alteraciones hidroelectrolíticas, uremia, trastornos tiroideos, entre otras)
E: Emotional (emocionales): pseudodemencia de la depresión
N: Nutrition (nutricional): deficiencia de vitamina B12, ácido fólico, entre otras.
T: Tumores y Traumas
I: Infections (infecciones): enfermedad de Lyme, sífilis, encefalitis, infección de vías urinarias, tuberculosis e intoxicaciones por plomo, talio y mercurio
A: Alcoholism (alcoholismo)
Es por esto también, que dentro del enfoque inicial del paciente con sospecha de demencia deben realizarse los siguientes paraclínicos: hemograma, electrolitos, pruebas de función renal, hepática y tiroidea, prueba de VIH, hemoglobina A1C, uroanálisis con sedimento, niveles de vitamina B12, pruebas para sífilis y una imagen cerebral (por lo común es una tomografía axial computarizada).
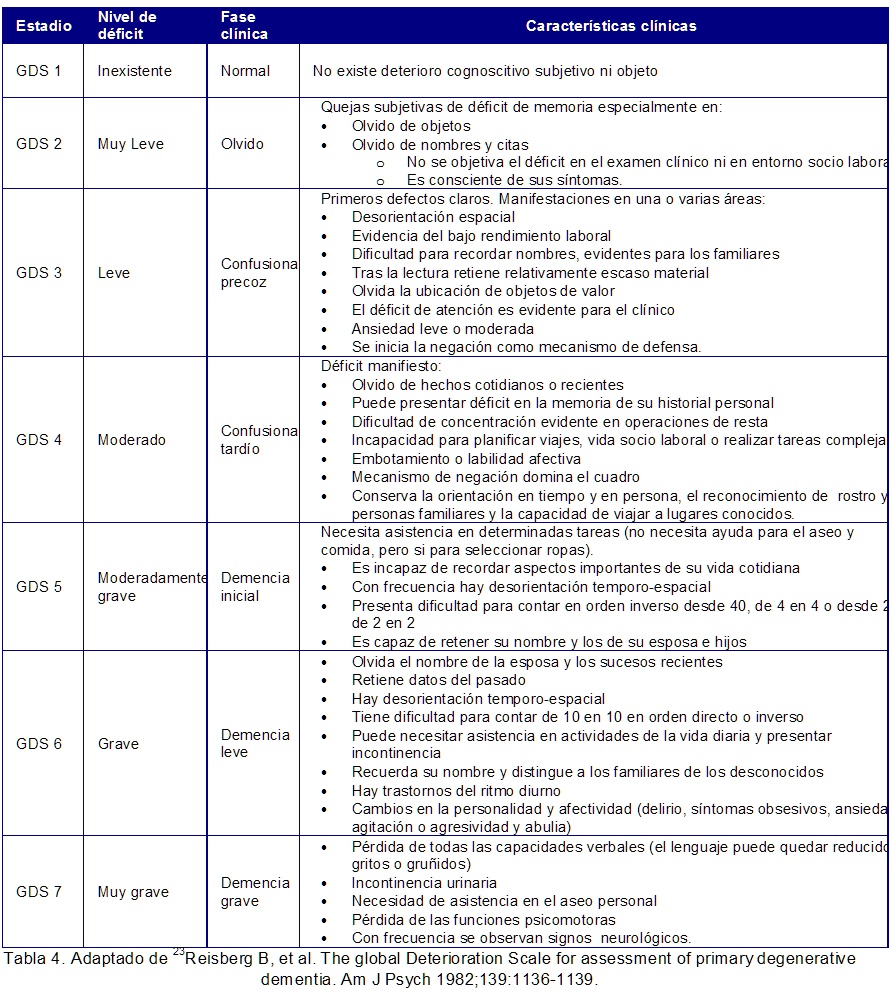
A algunos pacientes seleccionados se solicita pruebas reumatológicas, niveles de tiamina, medición del ácido metilmalónico, punción lumbar u otro tipo de imágenes cerebrales22. Una vez se ha realizado el diagnostico, se clasifica el estadio del paciente acorde con la escala GDS (Global Deterioration Scale), que tiene 7 estadios (Tabla 4)
Es importante recordar que el Minimental test, el Moca y la prueba del reloj (contenida en el Moca) nos dan información sobre la evolución clínica del paciente pero sobre todo nos dan indicios sobre la respuesta al tratamiento. En el capitulo "Dellirium y Deterioro cognoscitivo" encontraremos su rendimeinto diagnostico y de seguimiento.
Biomarcadores
Los diferentes biomarcadores que se han estudiado han sido principalmente dirigidos al estudio imagenologico y al líquido cerebroespinal24-26, en este útimo se han buscado 3 proteinas: la A42 que muestra los depósitos amiloides a nivel cortical; la proteína tau que refleja la intensidad de la neurodegeneración y la proteína tau fosforilada que se correlaciona con cambios neurofibrilares de la enfermedad27, tiene una sensibilidad y especificidad del 85% al 90% para identificar estadios preclínicos de Alzheimer en pacientes con deterioro cognoscitivo leve, pero su mayor relevancia tiene que ver con su buen valor predictivo negativo por lo que en un paciente con DCL con marcadores negativos casi que se podría excluir el diagnostico de EA5.
Se ha intentado medir estas mismas proteínas en sangre, sin embargo, ha sido complicado debido a las concentraciones tan bajas en sangre, a que estas proteínas son degradadas por proteasas plasmáticas y por la gran cantidad de proteínas de la matriz que causan interferencia en la medición, probablemente con los avances tecnológicos, podría llevarse a cabo esta medición5.
En cuanto a las imágenes, recordar que todo paciente debe tener una tomografía axial computarizada como parte del enfoque inicial de demencia. Las imágenes como marcador, se realizan principalmente a través de mediciones del volumen hipocampal incluyendo la segmentación del mismo para su evaluación. La resonancia magnética nuclear sigue siendo el método de elección para valorar cambios cerebrales vasculares como hiperintensidad de la sustancia blanca y micro sangrados (estos últimos están tomando gran valor ya que se han encontrado como reacciones adversas de los manejos anti A5.
Tratamiento
El tratamiento que tenemos en el momento para la EA es, muy a nuestro pesar, estrictamente sintomático, no se ha demostrado que ninguno de los tratamientos tenga un impacto claro en la evolución natural de la enfermedad, por lo que es fundamental en el momento de implementar los tratamientos, aclarar a los familiares las características del manejo para evitar expectativas no reales.
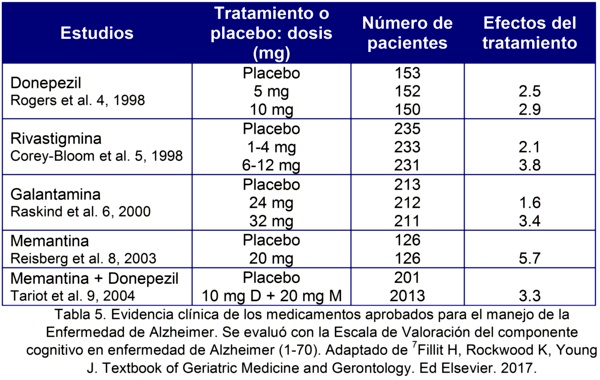
Inhibidores de la colinesterasa: como es sabido la disminución de la acetilcolina está asociada con DCL, delirium y demencia. Donepezil es el medicamento de este grupo con más selectividad tiene por la acetil-colinesterasa, la Rivastigmina inhibe la acetilcolinesterasa y la butiril-colinesterasa, por su parte la Galantamina no solo inhibe la acetil-colinestereasa, también modula los efectos de la acetilcolina en los receptores nicotínicos7.
Estudios doble ciego han demostrado que estos medicamentos aumentan la capacidad cognoscitiva, la capacidad en las actividades de la vida diaria y el comportamiento en pacientes con EA de leve a moderada, en escasa cantidad con manejo entre 6 y 18 meses. La Rivastigmina y el Donepezil han demostrado ser útiles en el manejo de la EA moderada a severa7 (Tabla 5).
Las dosis deben titularse para evitar las reacciones adversas (las cuales incrementan acorde con la dosis) siendo las mas comunes náuseas, vómito y diarrea.
El estudio AD 2000 32 y el Alzheimer´s Disease Cooperative Studies-Mild Cognitive Impairment Trial sugirieron que la efectividad del Donepezil (y los otros inhibidores de la colinesterasa) disminuye luego de 18 a 24 meses, sin embargo en el estudio AD 2000 se evidencian periodos de remisión, lo que haría pensar que probablemente no sea una declinación en la efectividad.
Medicamentos que impactan en el sistema glutamatergico: La Memantina antagoniza parcialmente el glutamato en los receptores NMDA (N-metil-D-aspartato) y actua de dos maneras:
Este grupo de medicamentos han demostrado mejoría de la capacidad cognoscitiva y comportamiento, en estadios moderado a severo. Con menor cantidad de efectos adversos.
Vitamina E: Anteriormente se consideraba que la reposicion con esta vitamina, retardaba el deterioro funcional, atención hospitalaria y muerte en 25% en pacientes con EA moderada a severa. El grupo de vitamina E del Alzheimer´s Disease Cooperative Study Group-Mild Cognitive Impairment no mostró beneficio comparado el placebo y en otros estudios la vitamina E a dosis de 2.000 unidades presentó mayor riesgo de trombosis.
TRASTORNO COGNOSCITIVO VASCULAR
El trastorno cognoscitivo vascular hace referencia a un grupo variable de manifestaciones en las cuales la causa primaria atribuible es la enfermedad cerebrovascular. Se reconoce como la principal causa de demencia después de la demencia tipo Alzheimer, se ha presentado un interés creciente en el rol de la lesión vascular cerebral en la patogénesis y manifestaciones clínicas de la enfermedad de Alzheimer. Estudios experimentales han mostrado un sinergismo funcional y patogénico entre endotelio vascular, las células neurales y la disfunción de la unidad neurovascular que intenta explicar el deterioro cognitivo. En la medida en que los factores de riesgo cardiovascular son modificables, el trastorno cognoscitivo vascular es una forma prevenible de demencia7.
El constructo del trastorno cognoscitivo vascular provee un abordaje amplio de deterioro cognitivo de origen vascular y va desde leve hasta muy severo; la patología de base es variable e incluye: infarto estratégico único, infartos múltiples, lesión de sustancia blanca no infartos como leucoaraiosis, hipoperfusión o hemorragia.
La prevalencia y la incidencia son variables, dependiendo de los criterios diagnósticos aplicados y de la población estudiada. La prevalencia de demencia vascular en estudios europeos realizados en los ańos 1990 reportó un incremento 0,3% en personas con edades entre los 65 ańos y 69 ańos, y un 5,2% en mayores de 90 ańos. La prevalencia de demencia vascular luego de ataque cerebrovascular va desde 6% hasta 32%, en reportes de 3 meses después de la condición aguda, es 3.5 a 5.6 veces más frecuente que en pacientes sin enfermedad cerebrovascular28.
Las variaciones geográficas en la prevalencia de trastorno cognitivo vascular se han demostrado, pero con datos aun no concluyentes, en algunos países del este de Asia se ha reportado mayor prevalencia para demencia vascular que para demencia Alzheimer, en las últimas tres décadas la proporción de demencia vascular a demencia Alzheimer ha cambiado de 2:1 a 1: 1 probablemente por el mejor control de los factores de riesgo vascular y prevención de enfermedad cerebrovascular29-30. Datos de otros países en vía de desarrollo reportan mayor prevalencia de demencia Alzheimer que vascular, pero se considera que la prevalencia de esta última si es mayor que en países industrializados.
Manifestaciones clínicas
No hay un único cuadro específico dado su heterogeneidad etiopatogénica y se plantea una clasificación por subtipos, a continuación:
a) De acuerdo a etiopatogenia
b) Síntomas psiquiátricos predominantes
Criterios diagnósticos
Como herramienta rápida para diagnóstico de demencia vascular se utiliza la escala isquémica de Hachinski con 13 ítems y con puntos de corte de 4 para DTA y 7 para demencia vascular con una sensibilidad de 89% y especificidad de 89%31. Recientemente se ha publicado una versión abreviada de escala de Hachinski con 7 ítems, con una aplicabilidad mejor que la versión inicial32.
Solo hasta hace poco, el termino trastorno neurocognitico vascular fue adoptado por el DSM-5 y trastorno cognitivo vascular por el grupo VASCOG. En el pasado se usó los criterios NINDS-AIREN, DSM-IV, CIE-10 con muy pobre asociación entre cada uno y difíciles de comparar.
Tratamiento
Los efectos del tratamiento son leves. Al revisar estudios y metanálisis se concluye que hay leves beneficios con el uso de inhibidores de colinesterasa y memantina33. Ensayos revisados en Cochrane concluyen que el donepecilo provee el mejor beneficio para deterioro cognitivo en demencia vascular pura y la galantamina para demencia mixta, los beneficios de rivastigmina y memantina no han sido probados34.
DEMENCIA POR CUERPOS DE LEWY
Demencias de Cuerpos de Lewy ha sido el nombre que se le ha asignado a un grupo de patologías, que comparten el hallazgo histopatológico postmortem de depósitos de alfa sinucleina a manera inclusiones citoplasmáticas eosinofílicas, de preferencia a nivel del tallo cerebral y en las neuronas dopaminérgicas de la sustancia nigra o a nivel frontotemporal en la corteza, las cuales pueden manifestarse bien sea como demencia de cuerpos de Lewy o demencia de la enfermedad de Parkinson. Dicho hallazgo histopatológico constituye, después de los hallazgos de amiloide y proteína Tau en la enfermedad de Alzheimer, el segundo más frecuente en pacientes con sospecha clínica de demencia en vida.
Fue en 1912 cuando por primera vez Frederick Lewy describió dichas inclusiones en pacientes con Enfermedad de Parkinson. En 1984 Kosaka y colaboradores notaron la predominancia de estas inclusiones frente a los ovillos neurofibrilares de proteína tau que se encuentran en la enfermedad de Alzheimer, en pacientes con enfermedad de Parkinson. Los aspectos claves para el diagnóstico fueron clarificados en 1996 en las guías publicadas por el primer consenso internacional del Consorcio de demencias de cuerpos de Lewy, actualmente no se cuenta con una imagen diagnostica ni un marcador bioquímico para su confirmación, pero el empleo y adaptación de estos criterios clínicos con el paso de los ańos ha demostrado ser útil, requiriendo, aun así, una mejora sustancial en su sensibilidad34.
Su forma de presentación es muy característica, pero dada la falta de familiaridad con la entidad en nuestro medio, tiene el riesgo de confundirse fácilmente con demencia tipo Alzheimer y con otras alteraciones como el delirium. Es además una patología para la que existen diversas propuestas terapéuticas destinadas a mejorar la calidad de vida de los pacientes y sus cuidadores, de ahí la importancia de su conocimiento. Esta revisión ofrece herramientas prácticas para su diagnóstico y manejo.
Epidemiología
La demencia de cuerpos de Lewy es el hallazgo histopatológico más frecuente en las demencias, excedido únicamente por la demencia tipo Alzheimer. En lo referente a la enfermedad de Parkinson y su relación con la demencia de cuerpos de Lewy se encuentra que en ambos el inicio antes de los 65 ańos no es común y ambos son más prevalentes en hombres que en mujeres.
A grosso modo, la prevalencia de demencia es del 25% en los pacientes con enfermedad de Parkinson y el riesgo de ésta se incrementa con la duración de la enfermedad, alcanzando el 50% a los 10 ańos después del diagnóstico. La incidencia es en términos generales de 100 por cada 1000 personas ańo, sin embargo, es mucho menor al inicio durante los primeros ańos del diagnóstico.
En cuanto a la demencia de cuerpos de Lewy como tal, hay tan solo algunos reportes de incidencia y prevalencia, una revisión sistemática estimó una proporción del 0 al 23% en los pacientes con demencia, con una prevalencia central del 4.2% con estudios basados en la comunidad y del 7.5% en estudios basados en clínica35.
Fisiopatología
Algunos investigadores creen que la demencia con Cuerpos de Lewy se relaciona poco con la enfermedad de Alzheimer y con la enfermedad de Parkinson, en una tendencia a considerar que es una entidad diferente que se localiza en el punto medio de las dos. Otros por su parte, dadas las múltiples características que comparten, conciben la posibilidad de que estas tres patologías sean diferentes fenotipos de un mismo proceso.
Clásicamente a los hallazgos fisiopatológicos de la enfermedad Alzheimer se han asociado placas de amiloide y ovillos neurofibrilares de proteína tau, más frecuentemente encontrados en las regiones parietal, temporal y parietoocipital de la corteza cerebral. Por su lado la enfermedad de Parkinson se asocia con cuerpos de Lewy que se observan de manera primaria en regiones subcorticales del cerebro, predominantemente en la sustancia nigra y el locus ceruleus. En contraste, la demencia de cuerpos de Lewy se caracteriza por la presencia de dichas inclusiones a nivel cortical, específicamente frontotemporal, con algunas localizaciones subcorticales, acompańándose además de placas de amiloide y ovillos neurofibrilares en algunas ocasiones, sin que necesariamente la clínica hable de una demencia mixta.
Sin embargo, aún se desconoce el papel que cumplen estas inclusiones citoplasmáticas en el cerebro, pues en algunos pacientes se ha encontrado abundancia del material sin síntomas, lo que ha llevado a considerar una posible función toxica versus una protectora; surgiendo así la hipótesis de una manera de producción multifactorial de la enfermedad, comprendida por factores que llevan a disminución de la reserva neural, encontrándose no solo depósitos proteicos en los cerebros de los pacientes, también perdida neuronal36.
Bioquímicamente la deficiencia de neurotransmisores también cumple un rol muy importante. La acetilcolina y la dopamina son los que se han visto más involucrados en el desarrollo de estas enfermedades, si bien en las tres se presencia una deficiencia considerable de cada uno, puede decirse que el predominio del déficit de acetilcolina ha caracterizado con mayor frecuencia a la enfermedad de Alzheimer, mientras las otras dos se han visto cualificadas por el déficit de dopamina.
Así pues, fisiopatológicamente han podido agruparse estas patologías en alfa sinucleinopatias cuando se trata de las demencias de Cuerpos de Lewy y se ha incluido a la enfermedad de Alzheimer dentro de las amiloidopatias.
Genética
Se sabe poco a cerca de la heredabilidad de la demencia de cuerpos de Lewy, por mucho tiempo se consideró como un trastorno esporádico dado su inicio tardío, falta de relaciones familiares con los pacientes y estudios no conclusivos con gemelos. Sin embargo, la fuerte relación que existe con la enfermedad de Alzheimer y la enfermedad de Parkinson, refuerzan la hipótesis de que la genética juega un rol importante en el desarrollo de la demencia de cuerpos de Lewy, la evidencia que soporta una posible predisposición genética proviene de que los estudios a familias con fenotipo mixto de demencia y parkinsonismo que han demostrado que estos cumplen un patrón de herencia mendeliana.
Cierto número de mutaciones genéticas han sido asociadas a la demencia de Cuerpos de Lewy y a la enfermedad de Parkinson, por ejemplo, actualmente se sabe que algunos errores genéticos parecen ser dosis dependiente, como el caso de la mutación o duplicación de alfa sinucleina que causa enfermedad de Parkinson autosómico dominante, pero la triplicación se asocia con frecuencia tanto a enfermedad de Parkinson y a demencia de Cuerpos de Lewy.
Otros genes también se relacionan con el riesgo de demencia de cuerpos de Lewy, el más prominente es GBA, el gen que codifica glucocerebrosidos. Mientras que las dobles mutaciones del GBA causan enfermedad de Gaucher (autosómico recesivo), una mutación simple se asocia con demencia de Cuerpos de Lewy junto con una variante de enfermedad de Parkinson que lleva a un mayor riesgo de deterioro cognitivo.
Por otro lado, se debe anotar que no todos los genes relacionados con la enfermedad de Parkinson confieren el riesgo de demencia, por ejemplo, las mutaciones del LRRK2, causan enfermedad de Parkinson autosómica dominante sin deterioro cognitivo.
Otro pequeńo número de genes han sido identificados como poseedores de riesgo para demencia de Cuerpos de Lewy, pero no para enfermedad de Parkinson, tales como el gen para la Apolipoproteina E (APOE), el gen MAPT que se ha asociado con taupatias y el COMT que se ha observado de manera variable en la enfermedad de Parkinson37.
Diagnostico
Como se dijo al inicio, al diagnóstico tanto de la demencia de cuerpos de Lewy como de la demencia de la enfermedad de Parkinson es sobre todo clínico. No existe aún un estándar de oro imaginologico o marcador bioquímico que permita su confirmación, esta se realiza únicamente postmortem con el estudio histopatológico del cerebro de los pacientes.
Así pues, el mayor reto diagnóstico es diferenciarlo de la enfermedad de Alzheimer en estadios tempranos de la enfermedad. El Consorcio de demencias de Cuerpos de Lewy viene presentando desde 1996 una tabla de criterios clínicos en los que se ofrece una combinación de criterios claves y criterios de soporte para hablar de un diagnostico probable y un diagnostico posible; a pesar de su baja sensibilidad, esta es la herramienta más útil con la que cuenta actualmente y ya se han puesto marcha diversos estudios que buscan incrementar la sensibilidad actual cercana al 32% sin sacrificar su especificidad del 95%, mediante una nueva comprensión de los criterios de soporte, tales como el comportamiento durante el sueńo con movimientos oculares rápidos38.
Otras estrategias ayudadoras que se han empleado hasta el momento para realizar el diagnostico presentan variados inconvenientes, por ejemplo, el reto con antipsicóticos es una actividad completamente desaconsejada, pues se relaciona con incremento de la mortalidad en estos pacientes. Otros recursos como el empleo de escalas de fluctuación, podrían ayudar, aunque debe tenerse en cuenta que éstas aún no se encuentran plenamente validadas, al igual que muchas pruebas neuropsiquiátricas.
La agudeza clínica es pues de suma importancia, especialmente al momento de diferenciar la demencia de Cuerpos de Lewy de la enfermedad de Alzheimer, sobre todo a sabiendas de que pueden existir las dos patologías en el mismo paciente.
Un acrónimo útil para recordar las características clínicas de la demencia de Cuerpos de Lewy es el siguiente:
DDaVP
Demencia: La demencia en los cuerpos de Lewy puede ser similar a la demencia en la enfermedad de Alzheimer, sin embargo los pacientes con cuerpos de Lewy tienden a tener mayores problemas con la función ejecutiva (planear, priorizar, secuenciar) y a tener alteraciones visoespaciales, aunque tienen mejor memoria verbal que los pacientes con Alzheimer, de tal manera que los resultados del Minimental test son más prominentes en los pacientes con Alzheimer, mientras las demencias con cuerpos de Lewy pueden tener hallazgos más valiosos en la prueba del reloj.
La demencia de los cuerpos de Lewy tiene una edad de presentación temprana, con declinación cognitiva progresiva que inicia hacia los 55 ańos. Dentro de los dominios también se observa la dificultad para la realización de multitareas y para seguir el hilo de una conversación. Con el tiempo las alteraciones cognoscitivas de los pacientes se incrementan hasta envolver varios dominios, cuando son suficientemente severas para alterar la función social u ocupacional, es decir, cuando impactan en las actividades de la vida diaria instrumentales o básicas, alcanzan el criterio para el diagnóstico de demencia.
Delirium (Estado cognitivo fluctuante - pseudodelirium): Ocurre en el 50% al 75% de los pacientes con demencia de Cuerpos de Lewy, las fluctuaciones pueden ocurrir en minutos horas o días y puede ser de mucha ayuda en diferenciarla de la enfermedad de Alzheimer. Este estado fluctuante imita al delirium, por lo que se le ha llamado pseudodelirium y contempla 4 aspectos claves en su determinación:
1. Mareo y letargia durante el día
2. Sueńo durante el día por dos horas o más
3. Permanencia en un solo sitio por mucho tiempo
4. Episodios de discurso desordenado
La presencia de 3 aspectos de los 4 se ve hasta en un 63% de los pacientes con demencia de cuerpos de Lewy y en un 12% de los pacientes con enfermedad de Alzheimer. Por ello, el estado cognoscitivo no puede valorarse solo con la primera visita al paciente, requiere de múltiples observaciones y el apoyo de la información provista por los familiares o cuidadores.
Alucinaciones Visuales: Los síntomas psicóticos ocurren en alrededor del 80% de los pacientes con demencia de cuerpos de Lewy, usualmente con alucinaciones puramente visuales, vívidas, a color, tridimensionales de humanos o animales y, aunque pueden ser perturbadoras para los cuidadores o familiares, por lo que general no son disfóricas o estresantes para los pacientes. También pueden ser ilusiones, lo que significa que presentan errores en la interpretación de los estímulos visuales, confundiendo por ejemplo un perchero con una persona. Es en este punto donde debe tenerse especial cuidado con la administración de medicación antipsicótica.
Parkinsonismo: En los pacientes con demencia de la enfermedad de Parkinson los síntomas motores preceden a la demencia varios ańos, mientras que en la demencia de Cuerpos de Lewy los síntomas motores aparecen al ańo de presentación de los síntomas cognitivos o pueden iniciar ambos grupos de síntomas de manera simultánea en estadios tempranos de la enfermedad.
Desde el punto de vista motor, la bradiquinesia, rigidez y caídas son lo más común en ambos tipos de demencia, sin embargo, el temblor está por lo general ausente en la demencia de cuerpos de Lewy. En estos pacientes con demencia de cuerpos de Lewy suelen presentar aumento de la base de sustentación, con mayor inestabilidad postural, dificultad para la marcha e inexpresividad facial. Los síntomas autonómicos como la hipotensión ortostática y la constipación son prominentes en la demencia de Cuerpos de Lewy y se presume que ambos responden bien al uso de levodopa/carbidopa para el control de los síntomas, pero la respuesta se encuentra un poco más limitada en la demencia de cuerpos de Lewy.
Otras características
Algunos autores incluyen la sensibilidad a los agentes antipsicóticos y el comportamiento durante el sueńo con movimientos oculares rápidos dentro de las características esenciales de la demencia de Cuerpos de Lewy, es más, esta última se ha propuesto como el mayor criterio diagnóstico, sin embargo, dentro del Consorcio Internacional, continúa siendo característica de apoyo.
La sensibilidad neuroléptica es resultado de la pérdida de neuronas dopaminérgicas, la administración de cualquier antipsicótico típico o altas dosis de atípicos, exacerba el parkinsonismo y en la enfermedad de Parkinson los cambios pueden ser irreversibles. Aquellos pacientes con demencia de cuerpos de Lewy son más sensibles a presentar un síndrome neuroléptico maligno, en términos generales son "alérgicos" al haloperidol y todos los antagonistas de los receptores D239.
El comportamiento durante el sueńo con movimientos oculares rápidos ocurre en cerca de la mitad de los pacientes y es con frecuencia el precursor en la demencia de cuerpos de Lewy al igual que un factor de riesgo para demencia de la enfermedad de Parkinson en los pacientes que ya poseen este diagnóstico. Este desorden se manifiesta por sueńos vívidos asociado con movimientos corporales simples o complejos durante el sueńo MOR, los pacientes actúan los sueńos, usualmente defendiéndose de ataques.
La incontinencia urinaria también es un síntoma característico en la demencia de Cuerpos de Lewy que puede ayudar a diferenciar de la enfermedad de Alzheimer, pues aparece en estadios tempranos. Otros aspectos como la depresión y los síntomas disautonómicos son comunes en la demencia.
Ayudas diagnósticas
Comparado con la enfermedad de Alzheimer, en donde biomarcadores de los diferentes tipos de amiloide en el líquido cerebroespinal han demostrado una buena sensibilidad y especificidad, la evidencia para la demencia de cuerpos de Lewy es insuficiente. Hay una clara necesidad por identificar la proteína alfa sinucleina para su empleo como biomarcador, sin embargo, algunos hallazgos séricos como por ejemplo el factor de crecimiento epidérmico puede tener cierta relevancia clínica al momento de predecir declinación cognitiva asociada a la enfermedad de Parkinson.
Imagenológicamente, tampoco se cuenta con hallazgos directos confirmatorios del diagnóstico, pero algunos aspectos pueden ser indicativos. Por ejemplo, la resonancia magnética no parece tener verdadera capacidad discriminatoria entre demencia de cuerpos de Lewy y enfermedad de Alzheimer, pero algunos estudios sugieren que la medición del volumen del putamen parece ofrecer orientación al momento de hacer el diagnóstico diferencial. Las imágenes provistas por la técnica PET SCAN pueden contribuir en la determinación de la distribución de la enfermedad, el hipometabolismo e hipoperfusión occipital es uno de los marcadores de demencia de Cuerpos de Lewy que soportan el diagnostico que podrían ayudar en el diagnóstico diferencial con la enfermedad de Alzheimer y enfermedad de Parkinson. Las imágenes realizadas con esta misma técnica que se enfocan en la determinación del transporte de dopamina particularmente a nivel de los ganglios basales, también ha demostrado cierta sensibilidad y especificidad, tanto así que el consenso del Consorcio de demencias de Cuerpos de Lewy, lo concibe como una característica muy sugestiva de la patología36.
Criterios diagnósticos para demencia de Cuerpos de Lewy
Los criterios del consenso del Consorcio internacional para demencias de cuerpos de Lewy, son un reflejo de las características clínicas ya descritas, se presentan a continuación:
Debe tenerse en cuenta que no toda enfermedad de Parkinson desemboca inevitablemente en una demencia, por eso tambien se habla de factores de riesgo para el desarrollo de demencia en enfermedad de Parkinson, entre ellos están:
La demencia de la enfermedad de Pakinson cuenta con caracteristicas clinicas similares a la demencia de Cuerpos de Lewy que pueden recogerse dentro de los criterios diagnosticos, el diagnostico diferencial como ya fue mencionado se encuentra en el tiempo de inicio desde el curso de los sintomas motores a los cognoscitivos.
DEGENERACIÓN LOBAR FRONTOTEMPORAL
Es un trastorno neurodegenerativo que afecta selectivamente el lóbulo frontal y temporal anterior resultando en fallas progresivas de lenguaje y cambios comportamentales7.
La demencia frontotemporal es la tercera causa de demencia neurodegenerativa, después de la DTA y demencia por cuerpos de Lewy40. Es una demencia de inicio temprano, pero al revisar reportes de casos de encuentra una variabilidad de presentación que va desde los 21 a los 89 ańos. Los datos epidemiológicos son escasos y los reportes de prevalencia e incidencia muy amplios, esto en parte a la heterogeneidad de sus manifestaciones clínicas, grupos poblacionales con diferente influencia genética y variabilidad de criterios diagnósticos41.
La incidencia de demencia frontotemporal en mayores de 65 ańos va de 3,44 a 16,7 pacientes por 100 000 pacientes ańo comparado con 0,64 a 4,1 por 100,000 pacientes ańos en el grupo de 64 ańos y menores42. Afecta hombres y mujeres por igual43, la duración de la enfermedad es más corta que la DTA, en promedio 6 a 7 ańos42. El principal factor de riesgo es la historia familiar positiva.
Presentación clínica
Existen dos variantes de demencia frontotemporal mejor reconocidas: variante comportamental caracterizada por cambios en la personalidad dados por comportamiento antisocial, pobre control de impulsos, embotamiento afectivo, conductas estereotipadas o ritualizadas. Se asocia con atrofia de corteza frontal anterior y anterotemporal44.
Los síntomas comportamentales son más severos cuando hay compromiso del hemisferio derecho. El trastorno cognitivo es menor relevante en sus etapas iniciales con mayor afectación de memoria de trabajo y función ejecutiva, dentro de los cambios cognitivos que se describen están alteración de la abstracción, atención, de la capacidad para resolver problemas, dificultad en la planeación, organización y perseveración45-46. Se reportan cambios dietaros principalmente por exceso en el consumo de dulces, glotonería que resultan en incremento de peso. La apatía se puede presentar con frecuencia y significa compromiso de la región cingulada anterior y frontal medial. Los trastornos del lenguaje se caracterizan por ecolalia, perseveración o mutismo y en el trascurso de la enfermedad se pueden presentar síntomas motores como parkinsonismo o síntomas de neurona motora41.
La variante afasia primaria progresiva se subdivide en variante semántica y variante agramática o afasia no fluente que se diferencian a continuación (Tabla 6).
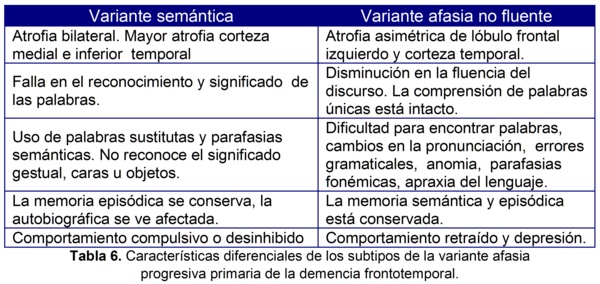
De las variantes de demencia frontotemporal la más frecuente es la comportamental, con una prevalencia de 57%, seguida de la afasia no fluente en un 24% y variante semántica en un 19% de los pacientes con este diagnóstico7.
Neuropatología
Se describe 3 fenotipos neuropatológicos
1. Acumulación anormal de micro túbulos hiperfosforilados asociado con proteína tau en células gliales y neuronales. Este subtipo se encuentra en menos del 50% de las autopsias47. Las principales patologías que tiene esta característica histopatológica son enfermedad de Pick, parálisis supranuclear progresiva, y degeneración cortico basal48.
2. Inclusiones neuronales ubiquitin reactivas y tau negativas40. En más del 50% de las autopsias. Proteína ligada a TAR DNA hiperfosforilada.
3. 5% a 10% autopsias. Inclusiones neuronales ubiquitin reactivas que consisten en sarcoma fusionado49.
Genética
El 40% de los pacientes con este diagnóstico tiene historia familiar positiva y de ellos el 20% a 30% exhibe un modo autosómico dominante50. La consanguinidad de primer grado tiene 3.5 veces más de riesgo de desarrollar demencia frontotemporal51. Se describen mutaciones en tres genes: progranulina52, cromosoma 953 y proteína tau asociada a micro túbulos54.
Diagnóstico
ABORDAJE DE PACIENTE CON DEMENCIA MODERADA A SEVERA
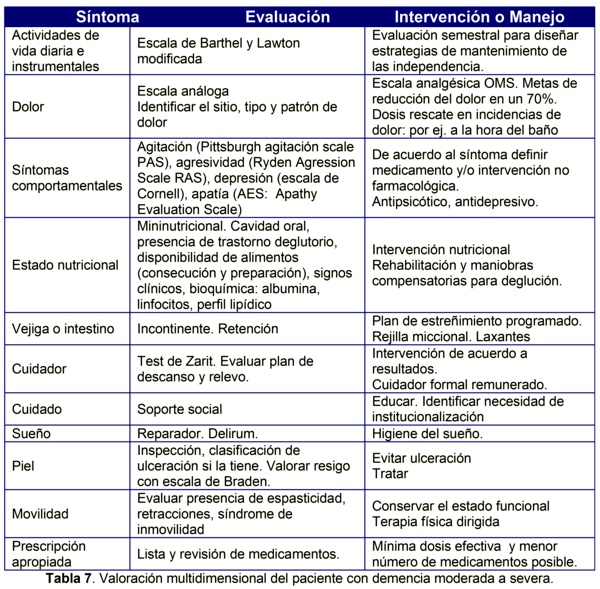
Tener el diagnóstico de demencia implica toda una serie de adaptaciones y cambios en el paciente y su familia; se plantea a continuación un abordaje geriátrico multidimensional especialmente en pacientes con mayor grado de deterioro cognitivo y funcional (Tabla 7).
ASPECTOS GENERALES DE TRATAMIENTO
El enfoque terapeutico se centra en el alivio sintomático de cada una de las caracteristicas que se van presentando con el objetivo de mejorar la calidad de vida, tanto de pacientes como de cuidadores; sin embargo, por esta misma razón el riesgo de incurrir en polifarmacia es alto y mas preocupante aún cuando la evidencia hasta el momento solo soporte el uso de los inhhibodres de la acetilcolinesterasa como medicamentos de uso optimo en esta patología. Aún así, se presentan recomendaciones terapeuticas ofrecidas por consenso de expertos, orientadas según grupo de sintomas:
REFERENCIAS
-
1. Diagnóstico y tratamiento integral de las demencias, capítulo 1, página 5. ISBN: 978-958-99088-3-9. 2013.
2. Classification and diagnosis of dementia: a mechanism- based approach. European Journal of Neurology 2009, 16: 168-173
3. Psiquiatría Geriátrica, Agűera Ortiz, Masson, 2002.
4. Alzheimer A: Uber einen eigenartigen schweren Krankheitsprozes der Hirnrinde. Neurologisches Centralblatt 25:113-114, 1906
5. Scheltens, Philip et al. Alzheimer´s disease. Lancet 2016; 388.10043:505-517.
6. Wu Y-T, Fratiglioni L, Matthews FE, et al. Dementia in western Europe: epidemiological evidence and implications for policy making. Lancet Neurol 2015;15:116-124
7. Fillit H, Rockwood K, Young J. Textbook of Geriatric Medicine and Gerontology. 8Ş edición. Ed Elsevier. 2017.
8. Brunkman AL, Goate AM: Presenilin function and gamma secretase activity. J Neurochem 93:769-792, 2005.
9. Jack CR Jr, Wiste HJ, Weigand SD, et al. Age,sex, and APOE e4 effects on Memory, brain structure, and beta-amyloid across the adult life span. JAMA Neurol 2015; 72:511-519
10. Bell RD, Winkler EA, Singh I, et al. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature 2012;485:512-516.
11. Rapp SR, Espeland MA, Shumaker SA, et al: Effect of estrogen plus progestin on global cognitive function in postmenopausal women. The Women's Health Initiative Memory Study: a randomized controlled trial. JAMA 289:2663-2672, 2003.
12. Yu F, Bronas U, Nelson N, et al: Aerobic exercise in Alzheimer's disease: the FIT-AD trial. Alzheimers Dement 10(Suppl ):P851- P852, 2014.
13. Rawlings AM, Sharrett A R, Schneider AL, et al. Diabetes in midlife and cognitive change over 20 years: a cohort Study. Ann Intern Med 2014;161:785-793.
14. De Bruijin RF, Bos MJ, Portegies ML, et al. The potential for prevention of dementia across two decades: the prospective, population-based Rotterdam Study. BMC Med 2015;13:132.
15. Filser S, Ovsepian SV, Masana M, et al. Pharmacological inhibition of BACE1 impairs synaptic plasticity and cognitive functions. Biol Psychiatry 2015;77:729-739
16. Vos SJB, Xiong C, Visser PJ, et al. Preclinical Alzheimer´s disease and its outcome: a longitudinal cohort Study. Lancet Neurol 2013;12:957-965.
17. Sperling RA, Aisen PS, Beckett LA: Toward defining the preclinical stages of Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement 2011;7:280-292.
18. McKhann GM, Knopman DS, Chertkow H: The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement 2011;7:263-269.
19. American Psychiatric Association: Diagnostic and statistical manual of mental disorders (DSM-V), ed 5, Washington DC, 2013, American Psychiatric Association.
20. Barnes J, Dickerson B, Frost C, Jiskoot LC, Wolk D, van der Flier WM. Alzheimer´s disease first symtoms are age dependent: evidence from the NACC data set. Alzheimers Dement 2015;11:1349-1357.
21. Aguera LF. Demencia y depresión: una interrelación multifactorial. Avances neurocientificos y realidad clínica (IV). Trastornos Cognitivos. Madrid: CYM, 2001:197-205.
22. Gomez JF, Curcio CL. Salud del anciano: valoración. Primera edición. Ed Blanecolor S.A.S. 2014
23. Reisberg B, Ferris SH, de León MJ, Crook T. The global Deterioration Scale for assessment of primary degenerative dementia. American Journal of Psychiatry 1982;139:1136-1139
24. Duits FH, Prins ND, Lemstra AW, et al. Diagnostic impact of CSF biomarkers for Alzheimer´s disease in a tertiary Memory clinic. Alzheimers Dement 2015;11:532-532.
25. O´Bryant SE, Gupta V, Henriksen K, et al. Guidelines for the standarization of preanalytic variables for blood-based biomarker studies in Alzheimer´s disease research. Alzheimers Dement 2015;11:549-63.
26. Blennow K, Dubois B, Fagan AM, Lewczuk P, de Leon MJ, Hampel H. Clinical utility of cerebrospinal fluid biomarkers in the diagnosis of early Alzheimer´s disease. Alzheimers Dement 2015;11:58-69.
27. Villemagne VL, Fodero-Tavoletti MT, Masters CL, et al: Tau imaging: early progress and future directions. Lancet Neurol 2015;14:114-124.
28. Lobo A, Launer LJ, Fratiglioni L, et al: Prevalence of dementia and major subtypes in Europe: a collaborative study of population-based cohorts. Neurologic Diseases in the Elderly Research Group. Neurology 54(Suppl 5):S4-S9, 2000.
29. Yoshitake T, Kiyohara Y, Kato I, et al: Incidence and risk factors of vascular dementia and Alzheimer's disease in a defined elderly Japanese population: the Hisayama Study. Neurology 1995;45:1161-1168.
30. Sekita A, Ninomiya T, Tanizaki Y, et al: Trends in prevalence of Alzheimer's disease and vascular dementia in a Japanese community: the Hisayama Study. Acta Psychiatr Scand 2010;122:319-325.
31. Moroney JT, Bagiella E, Desmond DW, et al: Meta-analysis of the Hachinski Ischemic Score in pathologically verified dementias. Neurology 1997;49:1096-1105.
32. Hachinski V, Oveisgharan S, Romney AK, et al: Optimizing the Hachinski Ischemic Scale. Arch Neurol 2012; 69:169-175.
33. Kavirajan H, Schneider LS: Efficacy and adverse effects of cholinesterase inhibitors and memantine in vascular dementia: a metaanalysis of randomised controlled trials. Lancet Neurol 2007;6:782-792.
34. Neef D, Walling AD. Dementia with Lewy bodies: an emerging disease. Am Fam Physician. 2006 Apr 1;73(7):1223-9.
35. Walker Z, Possin KL, Boeve BF, Aarsland D. Lewy body dementias. Lancet. 2015;386(10004):1683-1697.
36. Lippa CF, Duda JE, Grossman M, Hurtig HI, Aarsland D, Boeve BF, Brooks DJ, Dickson DW, Dubois B, Emre M, Fahn S, Farmer JM, Galasko D, Galvin JE, Goetz CG, Growdon JH, Gwinn-Hardy KA, Hardy J, Heutink P, Iwatsubo T, Kosaka K, Lee VM, Leverenz JB, Masliah E, McKeith IG, Nussbaum RL, Olanow CW, Ravina BM, Singleton AB, Tanner CM, Trojanowski JQ, Wszolek ZK; DLB/PDD Working Group. DLB and PDD boundary issues: diagnosis, treatment, molecular pathology, and biomarkers. Neurology. 2007;68:812-819.
37. Meeus B, Theuns J, Van Broeckhoven C. The genetics of dementia with Lewy bodies: what are we missing? Arch Neurol. 2012;69:1113-1118.
38. Gomperts SN. Lewy Body Dementias: Dementia With Lewy Bodies and Parkinson Disease Dementia. Continuum (Minneap Minn). 2016;22(2 Dementia):435-463.
39. Stinton C, McKeith I, Taylor JP, Lafortune L, Mioshi E, Mak E, Cambridge V, Mason J, Thomas A, O'Brien JT. Pharmacological Management of Lewy Body Dementia: A Systematic Review and Meta-Analysis. Am J Psychiatry. 2015;172(8):731-742.
40. Cairns NJ, Bigio EH, Mackenzie IR, et al: Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: consensus of the Consortium for Frontotemporal Lobar Degeneration. Acta Neuropathol 2007: 114:5-22.
41. Rosso SM, Donker KL, Baks T, et al: Frontotemporal dementia in The Netherlands: patient characteristics and prevalence estimates from a population-based study. Brain 2003;126:2016-2022.
42. Nilsson C, Landqvist WM, Nilsson K, et al: Age-related incidence and family history in frontotemporal dementia: data from the Swedish Dementia Registry. PLoS One 2014;9:94901.
43. Onyike CU, Diehl-Schmid J: The epidemiology of frontotemporal dementia. Int Rev Psychiatry 25:130-137, 2013.
44. Rascovsky K, Hodges JR, Knopman D, et al: Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain 2011;134:2456-2477.
45. Boxer AL, Boeve BF: Frontotemporal dementia treatment: current symptomatic therapies and implications of recent genetic, iochemical, and neuroimaging studies. Alzheimer Dis Assoc Disord 2007;21:S79-S87.
46. Neary D, Snowden J, Mann D: Frontotemporal dementia. Lancet Neurol 2005;4:771-780.
47. Sieben A, Van Langenhove T, Engelborghs S, et al: The genetics and neuropathology of frontotemporal lobar degeneration. Acta Neuropathol 2012;124:353-372.
48. Josephs KA, Hodges JR, Snowden JS, et al: Neuropathological background of phenotypical variability in frontotemporal dementia. Acta Neuropathol 2011;122:137-153.
49. Urwin H, Josephs KA, Rohrer JD, et al: FUS pathology defines the majority of tau- and TDP-43-negative frontotemporal lobar degeneration. Acta Neuropathol 2010;120:33-41.
50. Cruts M, Van Broeckhoven C: Loss of progranulin function in frontotemporal lobar degeneration. Trends Genet 2008; 24:186-194.
51. Stevens M, van Duijn CM, Kamphorst W, et al: Familial aggregation in frontotemporal dementia. Neurology 1998; 50:1541-1545.
52. Baker M, Mackenzie IR, Pickering-Brown SM, et al: Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature 2006;442:916-919.
53. De Jesus-Hernandez M, Mackenzie IR, Boeve BF, et al: Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011;72:245-256.
Comentario de la revisora Dra. Cynthia Marińansky . Geriatra de planta de la Unidad de Geriatria del Hospital Durand de Buenos Aires Directora de la Especialización en Geriatria Universidad Maimonides..
El presente artículo actualiza la temática de las demencias como marco conceptual y ángulo de diagnóstico diferencial con el delirium en el estudio de la incompetencia cognitiva.
Delirium y demencia constituyen un binomio que incentiva el ejercicio de una práctica clinica, analítica y focalizada en preservar la funcionalidad del paciente.
Comentario de la revisora Dra Silvina Dahl. Geriatra. Hogar Ledor Vador, Buenos Aires. Argentina
En la presente revisión se desarrolla, en forma extensa y actualiza, todo lo referido al diagnóstico, la clínica y los diferentes abordajes: psicofarmacológico y no-farmacológico de la alteración cognitiva del adulto mayor. Puede ser leído tanto por aquel que se inicia en el tratamiento de pacientes con demencia y delirium; así como por aquellos que necesitan o quieren mantenerse actualizados.