Indice del volumen Volume index
Comité Editorial Editorial Board
Comité Científico Scientific Committee
SÍNDROME DE GOOD DIAGNOSTICADO TRAS LA APARICIÓN DE APLASIA PURA DE SERIE ROJA Y TROMBOCITOPENIA AMEGACARIOCÍTICA ADQUIRIDA
María Victoria Cuevas, Ignacio Martínez-Sancho1, Teresa Lista2,
Ignacio Martínez-Cuevas3, Beatriz Cuevas.
Servicio de Hematología-Hemoterapia. 1Centro de Salud Las Huelgas.
2Servicio de Anatomía Patológica. 3Unidad de Investigación.
Hospital Universitario de Burgos. Burgos. Espańa
Email: mvcuevas @ hubu.es
Rev Electron Biomed / Electron J Biomed 2021;3: 18-25.
RESUMEN
El síndrome de Good es una forma de inmunodeficiencia adquirida caracterizada por la presencia de timoma con hipogammaglobulinemia, así como la ausencia de linfocitos B, linfopenia CD4+ e inversión del cociente CD4+/CD8+. Esta patología facilita la aparición de complicaciones infecciosas e inmunes
PALABRAS CLAVE: Síndrome de Good, Aplasia pura de serie roja, Trombocitopenia amegacariocítica.
ABSTRACT: Good's syndrome is a form of acquired immunodeficiency characterized by the presence of thymoma with hypogammaglobulinemia, as well as the absence of B lymphocytes, CD 4+ lymphopenia, and inversion of the CD4 + / CD8 + ratio. This pathology facilitates the appearance of infectious and immune complications
KEY WORDS: Good syndrome, Pure Red Cell Aplasia, Amegakaryocytic Thrombocytopenia
INTRODUCCIÓN
El síndrome de Good, descrito por Robert A. Good en 1954 es una inmunodeficiencia adquirida de inicio en la 4Ş o 5Ş década de la vida caracterizada por la presencia de timoma junto con hipogammaglobulinemia así como, ausencia de linfocitos B, linfopenia CD 4+ e inversión del cociente CD4+/CD8+. Esta patología facilita la aparición de complicaciones infecciosas e inmunes1.
CASO CLÍNICO:
Presentamos el caso de un paciente varón de 40 ańos de raza caucásica que como antecedentes refería haber presentado en enero/2019 diarrea crónica secundaria a infección por Campylobacter jeyuni con pérdida de 20 kilos de peso; en agosto/ 2019 había sido sometido a una timectomía (con histología de timoma subtipo B, estadio I de la clasificación de Masaoka sin miastenia).
En enero/2020, fue remitido a nuestro centro por hematuria detectándose anemia y trombocitopenia. En la exploración física no se objetivaron adenopatías ni visceromegalias, apreciándose hematomas dispersos.
Se realizaron los siguientes estudios:
En el hemograma aparecieron los siguientes valores: hemoglobina 11.5 gr/dl VCM 76 fl leucocitos 10,3 x 109/L, plaquetas 2 x 109/L, reticulocitos 0.2 % (5.4 x103/µl).
Las cifras de coagulación, Coombs D, bioquímica, metabolismo del hierro, vitamina B12, ácido fólico, hormonas tiroideas y complemento, fueron normales. Los anticuerpos anti-DNA, ANA, así como la serología de virus B, C y VIH fueron negativos.
La serología de citomegalovirus, virus Epstein-Barr y parvovirus B19 fue positiva para Ig G; la serología de sífilis fue negativa
El estudio de anticuerpos anticélulas parietales, antifactor intrínseco, anticardiolipina Ig G e Ig M, ANCA (MPO) ,antimicrosomales (A-TPO), ANCA (PR3), antirreceptor de acetilcolina, anti-receptor de TSH, anti-tiroglobulina arrojó un resultado negativo.
En el proteinograma tanto en suero como en orina no se apreciaba banda monoclonal y la determinación de Inmunoglobulinas mostró los siguientes valores: Ig A <27.9 mg/dL (70-400), Ig G 62.8 mg/dL (700.0-1600), Ig M <17.6 mg/dL (40.0-230); las subclases de Ig G presentaban cifras bajas excepto la IgG4.
El despistaje de hemoglobinuria paroxística nocturna fue negativo.
La citometría de flujo reveló una ausencia de Linfocitos B (CD19): 0%; Los Linfocitos T (CD3) fueron el 93,43% y el ratio CD4/CD8 de 1,23.
Para el estudio diagnóstico se realizó aspirado y biopsia de médula ósea; el aspirado de médula ósea reveló una médula ósea ligeramente hipercelular pero sin progenitores megacariocíticos ni eritroides (figura 1).
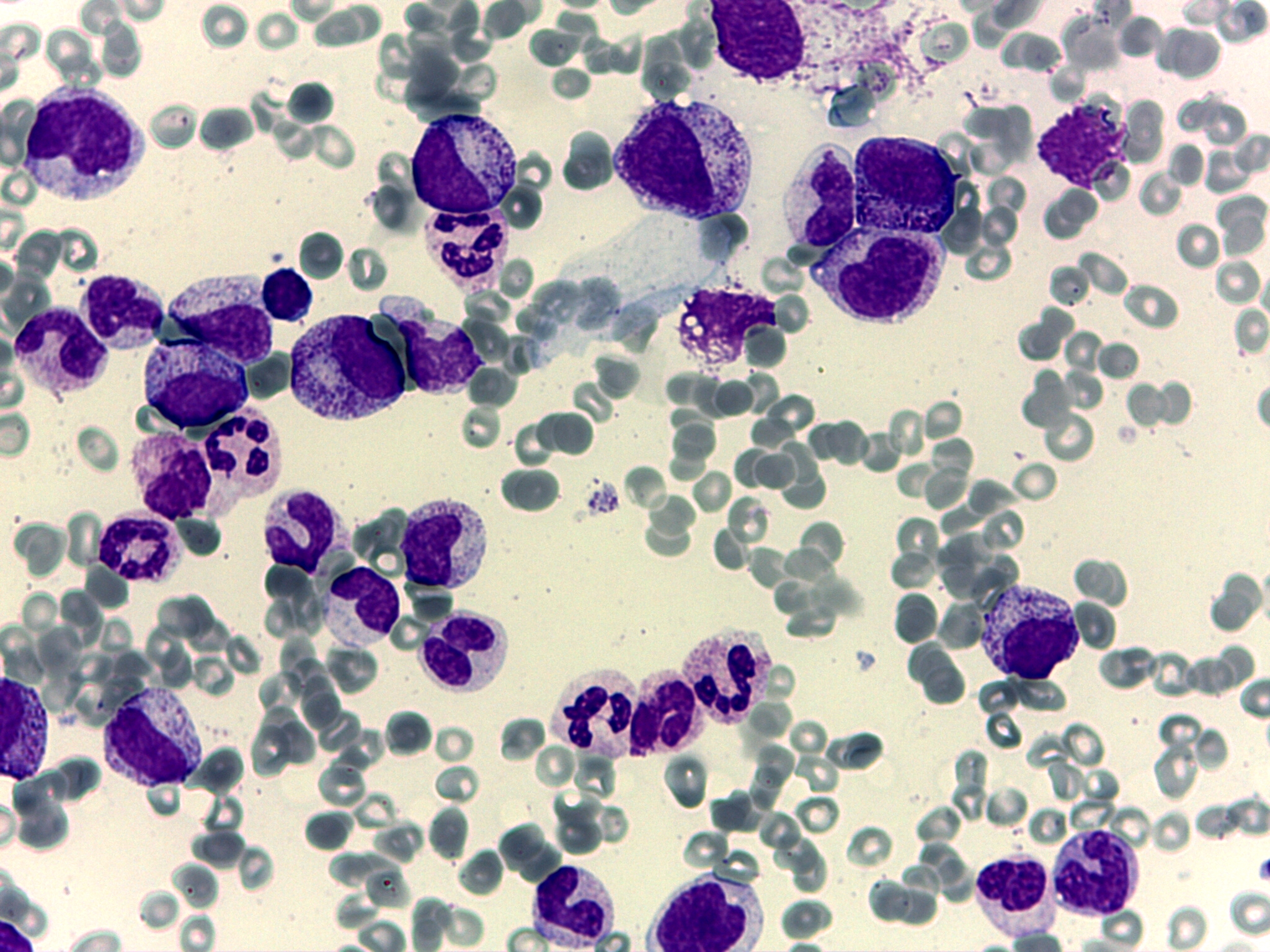
Tanto el cariotipo como el estudio de hibridación in situ (HIS) con sondas específicas de las regiones 5q, 7q, 8 y 20q fueron normales.
La biopsia ósea mostraba una celularidad del 70% constituida por serie granulocítica y linfocitaria; no se reconocían colonias de serie eritroide, ni megacariocitos, lo cual se confirmó con la negatividad para Glicoforina C y CD31/CD61 respectivamente. Se apreciaba hiperplasia de serie granulocítica con predominio de segmentados. Se observan frecuentes células mieloides precursoras paratrabeculares e intersticiales positivas para mieloperoxidasa y presencia de escasos blastos aislados positivos para CD34 en la zona centro medular. El CD117 fue positivo en células mieloides inmaduras paratrabeculares y también en algunos grupos centromedulares.
Con respecto a la serie linfoide se observaba un leve incremento del número de linfocitos T de aspecto maduro, intersticiales y en pequeńos agregados menores de 1 mm positivos para CD3 y a su vez positivos para CD4 y CD8 en proporción aparentemente habitual. TDT fue negativo. No se identificaron linfocitos B (PAX5, CD79a y CD20 negativos). Tampoco se observó fibrosis reticulínica ni colágena.
El diagnóstico fue compatible con Aplasia de serie roja y megacariocítica (figura 2).
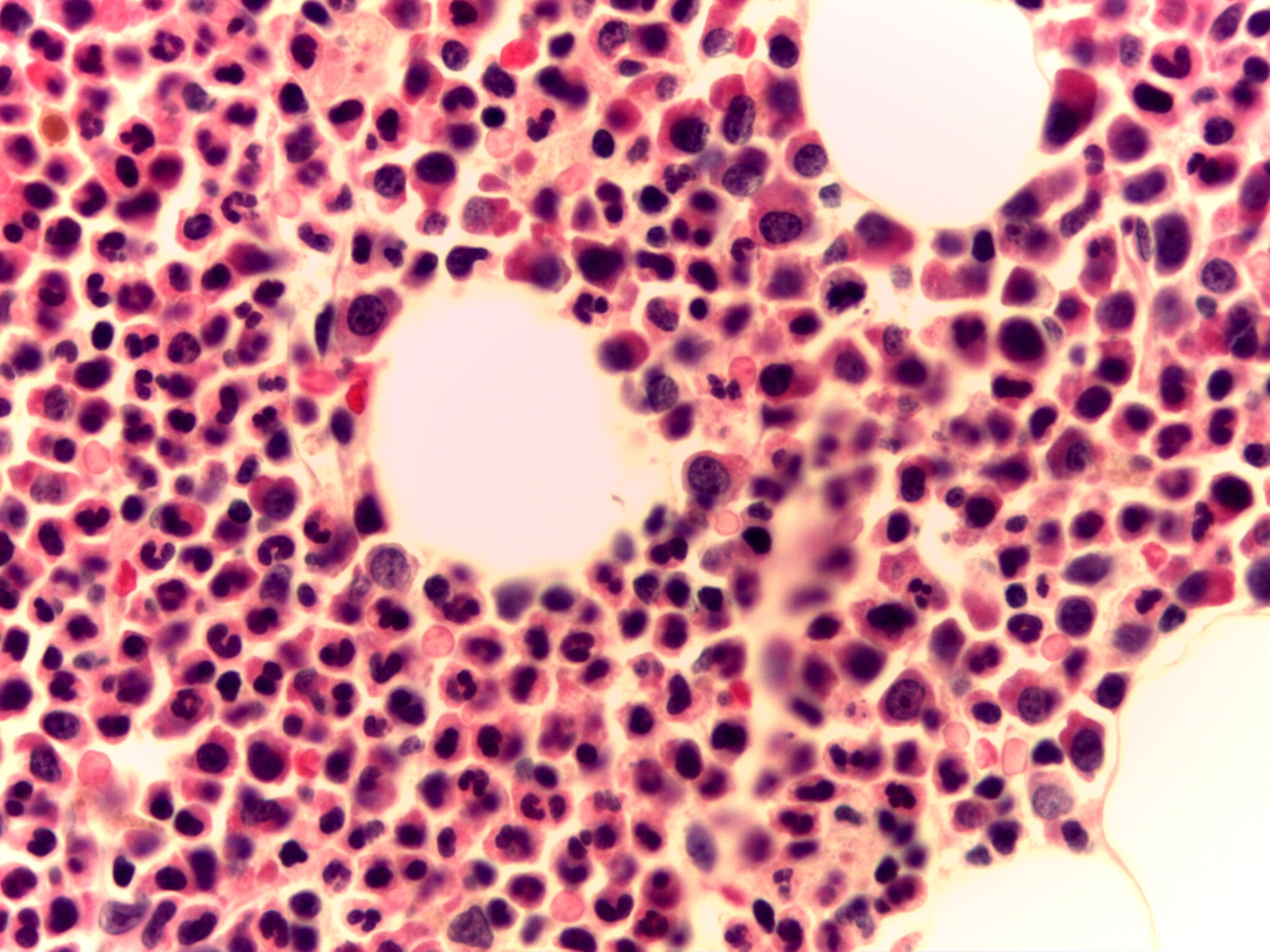
Para descartar patología tumoral o recidiva del timoma, se solicitó una tomografía computerizada (TC) que fue negativa y una Tomografía por emisión de positrones (PET) en la que no se evidenció patología metabólicamente activa.
Ante la sospecha del origen inmune de la anemia y la trombocitopenia, se inició tratamiento con Inmunoglobulina a la dosis de 1gr/kg/día durante 2 días y prednisona a dosis de 1 mg/kg.
Posteriormente, con los resultados de todas las pruebas y el diagnóstico de aplasia pura de serie roja (APSR) y trombocitopenia amegacariocítica adquirida (TAMA), el paciente recibió ciclosporina oral (para mantener niveles entre 200-400 ng/mL) y eltrombopag a dosis de 50 mg/día.
Siendo suspendido este último a los 14 días desde el inicio por normalización de la cifra plaquetas.
En las semanas posteriores, se procedió a disminuir la dosis de ciclosporina pues a los 92 días del inicio del cuadro se objetivó una cifra de hemoglobina normal. La ciclosporina fue suspendida a los 6 meses del debut de la aplasia pura de serie roja y la trombocitopenia amegacariocítica adquirida.
A los 6 meses se realizó una biopsia ósea en la que se apreciaba una recuperación tanto de la serie megacariocítica como eritroide (figura 3).
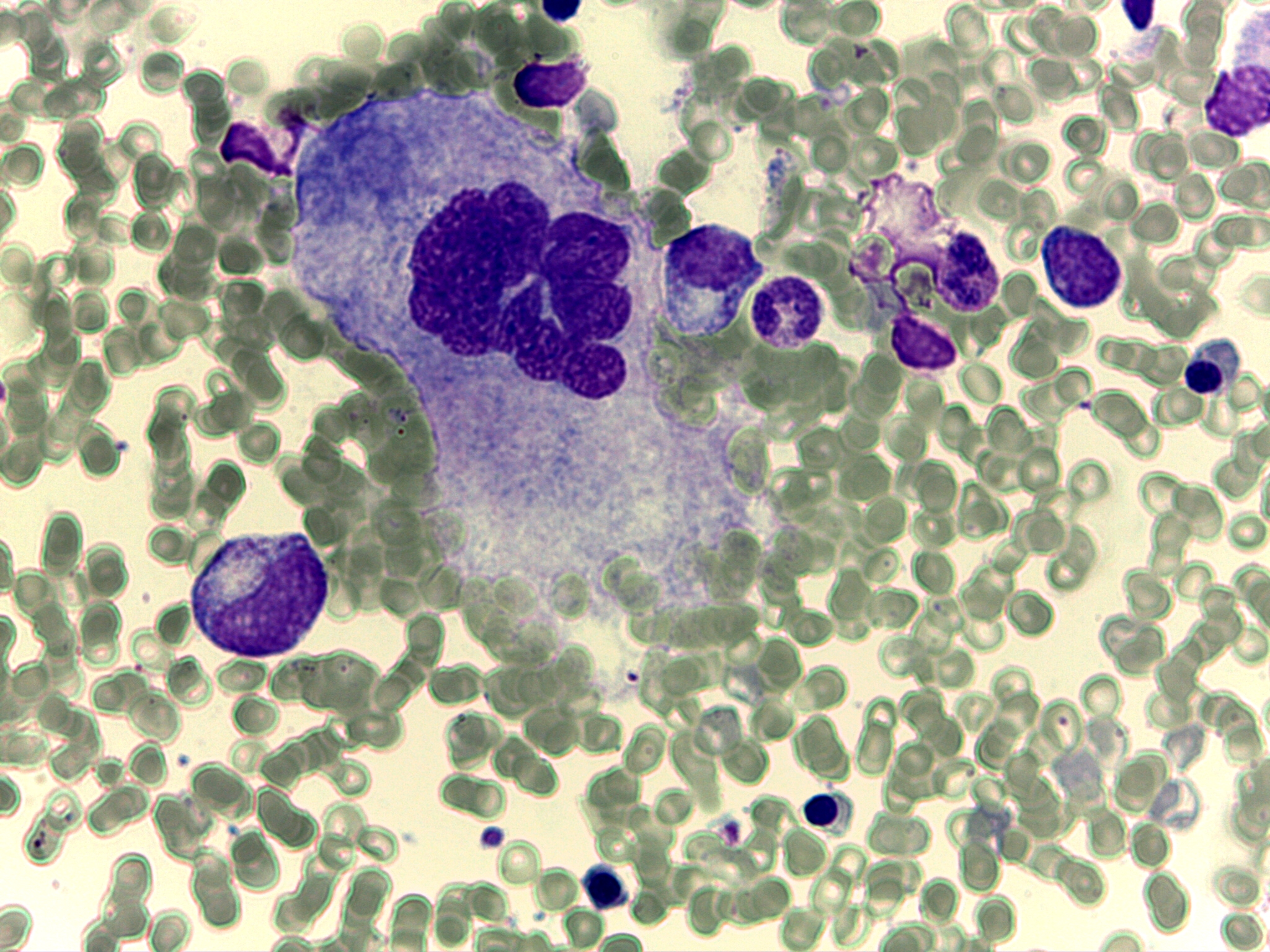
Tras 2 ańos de seguimiento el paciente no ha presentado complicaciones infecciosas o inmunes.
DISCUSIÓN:
El síndrome de Good es una forma de inmunodeficiencia adquirida descrita por primera vez por Robert A. Good en 1954 que se caracteriza por la presencia de timoma con hipogammaglobulinemia.
Esta asociación aparece en el adulto en la 4Ş o 5Ş década de la vida y los pacientes son susceptibles de sufrir infecciones bacterianas, víricas y fúngicas oportunistas debido a la deficiencia de la inmunidad humoral y celular que condiciona este síndrome.
Robert Alan Good fue un pionero en el campo de las inmunodeficiencias. Junto con sus colegas definió las bases celulares y las consecuencias funcionales de las inmunodeficiencias hereditarias. Fue uno de los grupos que descubrieron el papel fundamental del timo en el desarrollo de sistema inmune2
Los hallazgos inmunológicos descritos en el síndrome de Good incluyen el descenso de linfocitos B además de hipogammaglobulinemia, linfopenia CD4+ e inversión del cociente CD4+/CD8+ así como una reducción de la respuesta proliferativa a mitógenos por las células T.
Debido al escaso número de pacientes con esta patología, existen pocos estudios sobre la caracterización de las células del sistema inmune implicadas en la patogénesis y aunque se han postulado diferente hipótesis para explicarla, estas no son concluyentes.
El timo interviene en la actividad inmune principalmente en el desarrollo de las células T; tras migrar a la corteza tímica desde la médula ósea, los progenitores de células T son sometidos a una selección positiva o negativa asegurando el reconocimiento de antígenos extrańos y la tolerancia a los péptidos propios3.
Las células neoplásicas del timoma reemplazan a las células epiteliales normales y pueden causar disrupción de la maduración de las células T y por ello inducir la autotolerancia, que explicaría la propensión a desarrollar enfermedades autoimunes en el timoma4.
Tervasio et al., estudiaron la caracterización fenotípica de las células de sangre periférica por citometría de flujo de un paciente con este síndrome. Su estudio revela que además de la deficiencia de células B, hay una reducción del número de células dendríticas y de células CD8+ memoria; este menor número de células CD8+ memoria está involucrado en el incremento de la sensibilidad a las infecciones virales y también en la alteración del control inmunológico que provoca una cierta predisposición a la aparición de tumores.
En estos pacientes las infecciones por citomegalovirus y otros virus herpes son comunes; por ello, los autores sugieren que la alta predisposición a las infecciones por virus herpes y la reducción del control inmunológico de los tumores, podría ser el detonante de la aparición del timoma5
Aunque el síndrome tiene una distribución mundial, aproximadamente la mitad de los casos se han descrito en Europa.
Kelesidis y Yang hicieron una revisión y análisis exhaustivo de 152 casos publicados hasta 20106 y describen los siguientes hallazgos:
Con respecto a la edad de aparición, esta se sitúa entre los 40 y 70 ańos El diagnóstico de timoma precede a la hipogammaglobulinemia, la infección o diarrea en el 42 % de los casos. Sin embargo, hasta en el 38 % de los pacientes diagnosticados esta asociación fue simultánea o con una diferencia de 2 meses.
Con respecto a los tipos de histología del timoma según la clasificación de la OMS el más frecuente fue el tipo AB en el 41.7 % de los casos.
Los pacientes presentaron infecciones en un 82,3 % de los casos bacterianas, víricas, por micobacterias, fúngicas y por parásitos, siendo la principal causa de mortalidad.
En el 58.6% tuvieron manifestaciones autoinmunes siendo la más frecuente la aplasia pura de serie roja y en segundo lugar la miastenia gravis. Otras manifestaciones hematológicas descritas fueron: anemia hemolítica autoinmune, gammapatía monoclonal, síndrome mielodisplásico hemoglobinuria paroxística nocturna, aplasia pura de serie blanca, trombocitopenia y mielofibrosis.
La supervivencia media de estos pacientes es de 14 ańos y está reducida cuando se compara con la población control según edad y, no se ha observado que la supervivencia estuviera relacionada con el género, las enfermedades autoinmunes o el uso de inmunosupresores7
Con respecto a la aparición de APSR, se postula que sería debida a la destrucción de progenitores de serie roja mediados con células T y se ha visto en el 5 % de los pacientes con timoma.
La TAMA podría estar provocada por la disregulación tanto humoral como por la inmunidad mediada por células y consiste en la aparición de anticuerpos frente a la trombopoyetina y la destrucción de los megacariocitos por las células T.
La mayoría de los casos de TAMA no responden a corticoides o inmunoglobulinas endovenosas, sino a tratamiento inmunosupresor con ciclosporina, azatioprina o rituximab8
Onuki et al. describieron el hallazgo conjunto de aplasia pura de serie roja y trombocitopenia amegacariocítica y la detección posterior del timoma pero sin presencia de hipogammaglobulinemia siendo en este caso el tratamiento con ciclosporina eficaz y permitiendo la realización de timectomía9
El timoma ha sido asociado con menor frecuencia con la trombocitopenia amegacariocítica adquirida con solo 6 casos descritos en la literatura10
Por tanto, el síndrome de Good debe sospecharse en un paciente con antecedente de timoma que presenta hipogammaglobulinemia junto con complicaciones infecciosas y/o autoinmunes.
REFERENCIAS
-
1.- Good RA. Agammaglobulinemia- a provocative experiment of natura. Bull University Minnesota 1954; 26: 1-19.
2.- Ribatti D. The fundamental contribution of Robert A. Good to the discovery of the crucial role of thymus in mammalian immunity. Immunology. 2006; 119(3): 291-295.
3.- Shelly S, Agmon-Levin N, Altman A, Shoenfeld Y. Thymoma and autoimmunity. Cell Mol Immunol. 2011; 8(3): 199-202.
4.- Romi F. Thymoma in myasthenia gravis: from diagnosis to treatment. Autoimmune Dis. 2011; 2011: 474512.
5.- Ternavasio-de la Vega HG, Velasco-Tirado V, Pozo-Rosado L, Soler-Fernández MC, Pérez-Andrés M, Orfao A, Sánchez-Sánchez R, González-Villarón L. Persistence of immunological alterations after thymectomy in Good's syndrome: a clue to its pathogenesis. Cytometry B Clin Cytom. 2011; 80(5): 339-342.
6.- Kelesidis T, Yang O. Good's syndrome remains a mystery after 55 years: A systematic review of the scientific evidence. Clin Immunol. 2010; 135(3): 347-363.
7.- Jansen A, van Deuren M, Miller J, Litzman J, de Gracia J, Sáenz-Cuesta M, Szaflarska A, Martelius T, Takiguchi Y, Patel S, Misbah S, Simon A; Good syndrome Study Group. Prognosis of Good syndrome: mortality and morbidity of thymoma associated immunodeficiency in perspective. Clin Immunol. 2016; 171: 12-17.
8.- Dahal S, Sharma E, Dahal S, Shrestha B, Bhattarai B. Acquired Amegakaryocytic Thrombocytopenia and Pure Red Cell Aplasia in Thymoma. Case Rep Hematol. 2018; 2018: 5034741.
9.- Onuki T, Kiyoki Y, Ueda S, Yamaoka M, Shimizu S, Inagaki M. Invasive Thymoma with Pure Red Cell Aplasia and Amegakaryocytic Thrombocytopenia. Hematol Rep. 2016; 8(4): 6680.
10.- Simkins A, Maiti A, Short NJ, Jain N, Popat U, Patel KP, Oo TH. Acquired amegakaryocytic thrombocytopenia and red cell aplasia in a patient with thymoma progressing to aplastic anemia successfully treated with allogenic stem cell transplantation. Hematol Oncol Stem Cell Ther. 2019; 12(2): 115-118.
CORRESPONDENCIA:
Dra. María Victoria Cuevas
Servicio de Hematología-Hemoterapia.
Hospital Universitario de Burgos.
Avda. Islas Baleares 3.
09006 Burgos.
Espańa
Email: mvcuevas @ hubu.es
Recibido: 26/12/2021
Publicado: 30/12/2021